直接Z型Zn2SnO4-x N x/ZnO1-y N y異質結光催化劑的制備及機理
2019-09-09 08:07:44談國強穎黨明月任慧君傲劉
無機化學學報 2019年9期
王 敏 談國強*, 張 丹 李 斌 王 穎黨明月 任慧君 夏 傲劉 運
(1陜西科技大學材料科學與工程學院無機材料綠色制備與功能化陜西省重點實驗室,西安 710021)
(2陜西科技大學文理學院,西安 710021)
(3陜西科技大學電子信息與人工智能學院,西安 710021)
0 引 言
近幾年來,隨著世界范圍內經濟的快速發展和工業化步伐的加快,能源短缺和環境污染兩大問題日益突出,嚴重限制了人類社會的可持續發展。光催化技術作為一種環境友好、可持續發展的技術,可利用清潔可再生的太陽能和半導體基光催化劑同時解決以上2個問題[1-2]。在眾多傳統光催化劑中,TiO2因具有高熱穩定性、化學穩定性、高反應活性、低價等優點被廣泛研究[3]。但是,TiO2的禁帶寬度較大(~3.2 eV),僅能被占太陽能4%~5%的紫外光激發,并且光生電子-空穴對易復合,導致其量子產率較低,限制了其在光催化領域的進一步發展[4]。Zn2SnO4(ZTO)作為一種新型的三元氧化物半導體材料,具有高電子遷移率(10~15 cm2·V-1·s-1)、高電導率(104S·cm-1)和高熱穩定性等特點[5-6],在紫外光范圍內具有強光吸收特性,且在紫外光照射下可以降解甲基橙、亞甲基藍、羅丹明B、苯酚、甲醛和一氧化碳等多種有毒物質。但是,與TiO2類似,ZTO也是一種寬禁帶半導體,其禁帶寬度約為3.7 eV,對太陽光的利用率極低。
金屬元素摻雜可應用于ZTO基光催化劑的改性研究[7],雜質元素的存在可以通過調控其電子結構和光學性質從而提高其光催化活性。但是金屬元素摻雜會降低半導體的穩定性,并且光腐蝕現象的存在會使金屬元素溶出,從而造成二次污染[8]。因此,非金屬元素摻雜成為半導體基光催化劑改性的首選方法之一。向光催化劑中引入非金屬雜質元素可以通過形成雜質能級拓寬其光響應范圍,從而提高其光催化性能[9-10]。但是,非金屬摻雜同時會向半導體中引入大量的缺陷和表面態,成為光生電子-空穴對的復合中心,對其光催化性能的提高起反作用[11-12]。此外,與能帶結構相匹配的半導體復合,例如SnO2[13]、BiOI[14]、BiOBr[15]、V2O5[16]、g-C3N4[17]等 , 構 筑 異質結,可以通過提高光生載流子的分離效率從而提高ZTO的光催化性能。但是,對于傳統的異質結,載流子在界面處分離的同時會導致光生電子和空穴的氧化還原能力削弱,反應活性降低。構筑新型的Z型異質結可以解決這一問題[18-19]。
結合以上2種改性方法的優缺點,采用微波溶劑熱法一步合成N摻雜Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑,通過在紫外光照射下降解羅丹明B(RhB)、亞甲基藍(MB),甲基橙(MO)和水楊酸(SA)等污染物表征其光催化活性,并通過活性物種捕獲實驗驗證異質結構的類型,為制備新型Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑提供研究思路。
1 實驗部分
1.1 Zn2SnO4-x N x/ZnO1-y N y光催化劑的制備
稱取0.595 g Zn(NO3)2·6H2O加入到20 mL乙二醇中,磁力攪拌20 min后得到濃度為0.1 mol·L-1的硝酸鋅溶液;稱取0.351 g SnCl4·5H2O加入到20 mL去離子水中,磁力攪拌20 min后得到濃度為0.05 mol·L-1的氯化錫溶液;然后將氯化錫水溶液緩慢加入到硝酸鋅的乙二醇溶液中,磁力攪拌20 min將溶液混合均勻;再向上述混合溶液中加入一定量0.6 mol·L-1的 N2H4·H2O 溶液,磁力攪拌 30 min 后得到乳白色的前驅液。將所得前驅液轉移到內襯為聚四氟乙烯的微波水熱反應釜中,200℃下反應60 min后自然冷卻至室溫。收集產物,用去離子水和無水乙醇分別洗滌3次后,70℃下恒溫干燥12 h即得Zn2SnO4-xNx和Zn2SnO4-xNx/ZnO1-yNy復合光催化劑。當前驅液中0.6 mol·L-1N2H4·H2O溶液的加入量分別為 14、15、16、17、18、19 和 20 mL 時,所得前驅液中 N2H4·H2O 的 濃 度 分 別 為 0.156、0.164、0.171、0.179、0.186、0.193 和 0.200 mol·L-1, 結合后文表征結果,將所得產物分別命名為ZTN1、ZTN2、ZTN/ZN1、ZTN/ZN2、ZTN/ZN3、ZTN/ZN4 和 ZTN/ZN5。
ZnO1-yNy的 制 備 過 程 如 下 :0.595 g Zn(NO3)2·6H2O溶解到20 mL乙二醇和20 mL去離子水的混合溶液中,混合均勻后加入18 mL 0.6 mol·L-1的水合肼溶液,200℃下反應60 min后冷卻、洗滌、干燥,得到ZnO1-yNy粉體。
1.2 Zn2SnO4-x N x/ZnO1-y N y光催化劑表征
采用 X射線衍射儀(XRD,D/max-2200 PC,Rigaku,Cu Kα,λ=0.154 06 nm,40 kV,40 mA) 對樣品的物相組成和晶體結構進行表征,掃描速率為7°·min-1,掃描范圍為 10°~70°。 樣品的光吸收特性通過紫外-可見漫反射圖(UV-Vis DRS,Cray 5000,Agilent,U.S.A.)反映,測試范圍為 200~600 nm。 使用透射電子顯微鏡(TEM,FEI-Tecnai G2 F20)觀察樣品的表面形貌、組成和微觀結構,其工作電壓為300 kV。
1.3 Zn2SnO4-x N x/ZnO1-y N y光催化實驗
通過在紫外光照射下降解RhB評估樣品的光催化活性,300 W汞燈被用作紫外光光源。0.05 g光催化劑加入到50 mL初始濃度為5 mg·L-1的RhB水溶液中,置于XPA-7光化學反應儀(南京胥江機電廠)中,在磁力攪拌作用下暗反應30 min,待光催化劑粉體與染料分子間達到吸附-脫附平衡后開始光照,每隔一定時間取5 mL懸濁液,離心分離(3 500 r·min-1)取上清液,用紫外-可見分光光度計(SP-756P)測試其在554 nm處的吸光度(A)的變化。根據吸光度的改變計算RhB的降解率η:
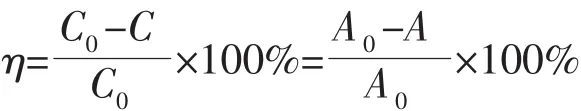
式中:C0為RhB溶液的初始濃度,C為光催化反應過程中某一時刻RhB溶液的濃度;A0為初始的RhB溶液在554 nm處的吸光度,A為光催化反應過程中某一時刻RhB溶液在554 nm處的吸光度。MB、MO和SA的降解反應與RhB類似,不同的是,以上3種污染物水溶液的初始濃度為10 mg·L-1。
1.4 Zn2SnO4-x N x/ZnO1-y N y光催化劑光電化學測試
使用CHI-660E電化學工作站對樣品的光電化學性能進行表征。測試系統為標準三電極體系:Pt片作為對電極,飽和Ag/AgCl電極為參比電極,涂覆有樣品的氟摻雜氧化錫(FTO)導電玻璃為工作電極,0.1 mol·L-1的Na2SO4溶液為電解液。工作電極的制備過程如下:稱取0.02 g光催化劑粉體分散到1 mL無水乙醇和0.2 mL乙酰丙酮的混合溶液中,超聲處理30 min后得到分散均勻的懸濁液。將該懸濁液旋涂到1.5 cm×2 cm的FTO玻璃基板上,在馬弗爐內200℃退火2 h后隨爐冷卻至室溫即得工作電極。在紫外光照射下測試樣品的瞬態光電流響應(I-t曲線),測試時間為400 s,無外加偏壓,開關燈間隔均為2 s;在紫外光照射下測試樣品的電化學阻抗圖譜(EIS),所選頻率范圍為 0.01~1×105Hz。 300 W 汞燈被用作紫外光光源。Mott-Schottky測試在暗光條件下進行,測試范圍為-1~1 V。使用ZSimpWin軟件對樣品的Nyquist點圖進行擬合。
2 結果與討論
2.1 Zn2SnO4-x N x/ZnO1-y N y的組成與晶體結構分析
圖 1a為樣品的 XRD 圖。 在 2θ=17.86°、29.15°、34.33°、41.70°、55.07°和 60.44°處的衍射峰分別對應立方相反尖晶石結構Zn2SnO4(PDF No.24-1470)的(111)、(220)、(311)、(400)、(511)和(440)晶面,而 2θ=31.77°、36.25°和 56.59°處的衍射峰則分別對應六方相 ZnO(PDF No.36-1451)的(100)、(101)和(110)晶面,說明 ZTN/ZN1、ZTN/ZN2、ZTN/ZN3、ZTN/ZN4 和 ZTN/ZN5樣品中包含Zn2SnO4和ZnO兩種物相。當前驅液中N2H4·H2O濃度較低時,樣品ZTN1和ZTN2未出現ZnO相。隨著N2H4·H2O濃度的增大,樣品的結晶度先增大后減小,但其他樣品的結晶度均大于ZTN1和ZTN2,說明堿性條件有利于ZnO的生成和樣品結晶度的提高。較低結晶度的樣品,意味著其具有較大的表面缺陷濃度和表面能。圖1b是樣品的XRD 圖在 30°~40°范圍內的放大圖。 ZTN/ZN1、ZTN/ZN2、ZTN/ZN3、ZTN/ZN4 和 ZTN/ZN5 中 的 Zn2SnO4(311)晶面的衍射峰發生了不同程度的偏移,說明Zn2SnO4與ZnO之間存在相互作用力,即形成了異質結[20]。此外,與標準卡片相比,Zn2SnO4的(311)晶面以及ZnO的(101)晶面的衍射峰產生的明顯偏移是由N摻雜所引起的晶格畸變導致的,說明N元素被成功的引入到了Zn2SnO4和ZnO的晶格中,形成了Zn2SnO4-xNx/ZnO1-yNy晶體。
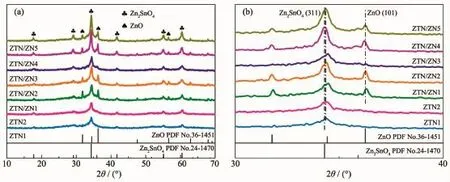
圖1 (a)樣品的XRD圖;(b)XRD放大圖Fig.1 (a)XRD patterns of samples;(b)Magnified XRD patterns
2.2 Zn2SnO4-x N x/ZnO1-y N y的形貌和元素組成分析
圖 2(a,b)分別是 ZTN1和 ZTN/ZN3號樣品的TEM圖。圖2a表明制備的Zn2SnO4由不規則納米顆粒組成,顆粒尺寸較小,并且表現出明顯的團聚現象。從圖2b可以看出,ZTN/ZN3號樣品由2種不同的形貌組成,Zn2SnO4為不規則納米顆粒,而ZnO則表現為納米棒形貌。Zn2SnO4納米顆粒緊密地附著在ZnO納米棒上,形成類核殼結構的界面異質結。圖2c是ZTN/ZN3號樣品的高分辨率透射電子電鏡(HRTEM)圖,晶面間距 0.310、0.264和 0.291 nm 分別對應于立方相反尖晶石結構 Zn2SnO4的(220)、(311)晶面和六方相ZnO的(100)晶面,證明Zn2SnO4和ZnO兩相在復合材料中共存。圖2d為選區電子衍射(SAED)圖,環形的衍射斑點說明Zn2SnO4為多晶結構,ZnO為單晶結構。圖2(e~h)為ZTN/ZN3的元素分布圖,除了Zn、Sn、O元素之外,N元素同樣可以被探測到,說明N元素被成功地引入到了Zn2SnO4和ZnO的晶格中,形成了Zn2SnO4-xNx和ZnO1-yNy結構單元,表明以N2H4·H2O為N源,采用微波溶劑熱法成功制備了N摻雜Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑。
基于以上分析和相關文獻報道[21-25]得出Zn2SnO4-xNx/ZnO1-yNy異質結構的形成機理。Zn(NO3)2·6H2O和SnCl4·5H2O在乙二醇和去離子水中解離成相應的離子,形成 2個透明溶液(反應式(1~3),圖 3a)。將SnCl4·5H2O的水溶液分散到Zn(NO3)2·6H2O的乙二醇溶液后形成ZnSn(OH)6和Zn(OH)42-膠核(反應式(4,5))。加入水合肼溶液,攪拌均勻得到反應前驅液(圖3(b,c))。 由于N2H4·H2O在去離子水中解離形成N2H5+和OH-(反應式(6))。在靜電引力作用下,溶液中的 N2H5+離子吸附在 ZnSn(OH)6和Zn(OH)42-膠核表面,形成 ZnSn(OH)6·N2H5+和 Zn(OH)4(N2H5)2膠粒(反應式(7))。水熱反應后,N雜質原子分別進入Zn2SnO4和ZnO的晶格中,形成Zn2SnO4-xNx和ZnO1-yNy(反應式(8),圖 3d)。 N 原子在 Zn2SnO4-xNx和 ZnO1-yNy的價帶頂部形成雜質能級,增大了Zn2SnO4和ZnO表面電荷密度,從而導致晶格畸變和界面變化。
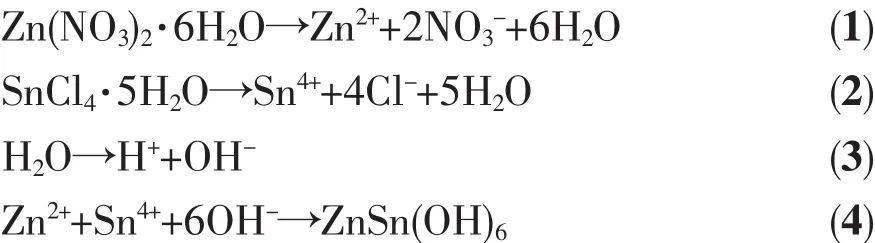

圖2 (a)ZTN1和(b)ZTN/ZN3的TEM圖;ZTN/ZN3的(c)HRTEM圖和(d)SAED圖;ZTN/ZN3 中(e)Zn、(f)Sn、(g)O 和(h)N 的 TEM mapping圖Fig.2 TEM images of(a)ZTN1 and(b)ZTN/ZN3;(c)HRTEM and(d)SAED images of ZTN/ZN3;TEM mapping images of(e)Zn,(f)Sn,(g)O and(h)N in ZTN/ZN3
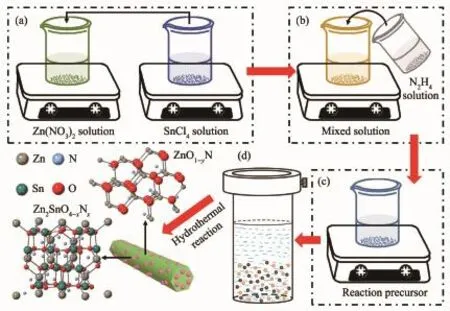
圖3 Zn2SnO4-x N x/ZnO1-y N y異質結的形成機理Fig.3 Formation mechanism of Zn2SnO4-x N x/ZnO1-y N y heterojunction
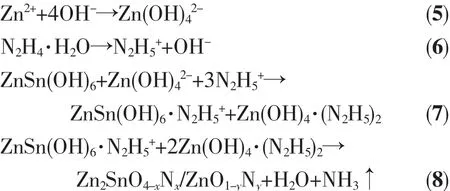
2.3 Zn2SnO4-x N x/ZnO1-y N y的紫外-可見漫反射光譜分析
圖4是光催化劑的紫外-可見漫反射(UV-Vis DRS)光譜,ZTN1和ZTN2在200~300 nm范圍光吸收明顯大于其他光催化劑,是因為ZnO1-yNy的存在降低了其他樣品在紫外光范圍內的光吸收強度。另外兩者在350~400 nm范圍內微弱的吸收肩源于異質元素摻雜[26-27],進一步說明N原子取代O原子在Zn2SnO4價帶頂部形成N雜質能級。對于ZTN/ZN1、ZTN/ZN2、ZTN/ZN3、ZTN/ZN4 和 ZTN/ZN5, 在 280~320 nm范圍內的光吸收邊帶歸屬于Zn2SnO4-xNx,而400 nm左右的光吸收邊帶的出現則歸因于ZnO1-yNy的存在。這進一步證明Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑被成功合成。
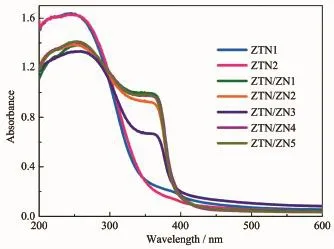
圖4 樣品的紫外-可見漫反射光譜Fig.4 UV-Vis DRS of samples
2.4 Zn2SnO4-x N x/ZnO1-y N y的光催化活性和穩定性分析
圖5 a為光催化劑在紫外光照射下對RhB的降解曲線。ZTN1和ZTN2在紫外光照射30 min后對RhB的降解率分別為90.63%和86.05%。而ZTN/ZN3在30 min內對RhB的降解率為94.56%,說明Zn2SnO4-xNx與ZnO1-yNy形成異質結之后,有效地提高了Zn2SnO4-xNx的光催化活性。而其他異質結光催化劑樣品光催化性能略低于ZTN1和ZTN2樣品,這是由ZnO1-yNy的光腐蝕現象導致的[28-29]。當異質結中ZnO1-yNy的含量較大時,暴露在外的ZnO1-yNy增多,光腐蝕現象的增強導致光催化性能降低,而適量匹配的ZTN/ZN3樣品形成Zn2SnO4-xNx納米顆粒緊密地附著在ZnO1-yNy納米棒上的ZnO@Zn2SnO4核殼結構的界面異質結,弱化了ZnO1-yNy的光腐蝕現象。根據Langmuir-Hinshelwood模型,RhB的光催化降解過程為偽一級反應。其表觀速率常數可以根據經驗動力學方程(9)計算[30]:

式中,C0′、C、kapp和 t分別代表暗反應吸附平衡之后RhB溶液的濃度、光催化過程中某一時刻RhB的濃度、表觀速率常數(min-1)和光照時間(min)。根據上述公式計算得到所有樣品的表觀速率常數如圖5b所示。ZTN/ZN3具有最大的表觀速率常數(0.097 0 min-1),分別為 ZTN1(0.069 2 min-1)和 ZTN2(0.067 6 min-1)的1.40倍和1.43倍。該結果表明Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑在紫外光照射下的光催化活性較純相Zn2SnO4-xNx有所提高。其性能提高的原因是ZnO1-yNy@Zn2SnO4-xNx核殼結構的界面異質結及雙N雜質能級的效應拓寬了光響應范圍。
為考察Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑的循環穩定性,以ZTN/ZN3作為光催化劑,經過5次循環實驗之后(圖6a),樣品的光催化活性并沒有明顯降低,在第5次循環降解實驗中,紫外光照射40 min后,RhB的降解率仍可達96.03%;圖6b為反應前后ZTN/ZN3的XRD圖,反應前后樣品的物相結構沒有明顯改變,其衍射峰強度的降低可以歸因于在水相條件下,部分小分子吸附在樣品表面,導致其XRD衍射峰強度降低。以上結果表明Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑具有良好的循環穩定性。
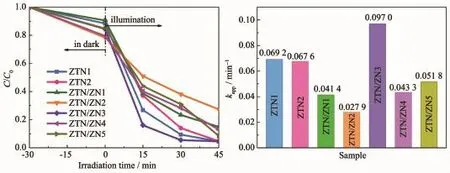
圖5 (a)樣品對RhB的光催化降解曲線;(b)表觀速率常數Fig.5 (a)Photocatalytic degradation curves of RhB over samples;(b)Apparent rate constants
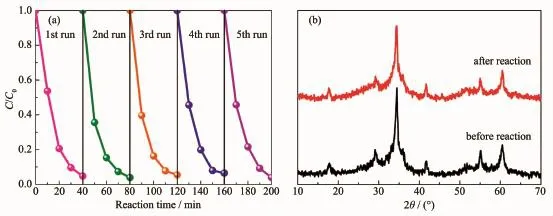
圖6 (a)ZTN/ZN3在紫外光照射下對RhB的循環降解實驗;(b)循環反應前后ZTN/ZN3的XRD圖Fig.6 Recycle degradation experiments of RhB over ZTN/ZN3 under UV light irradiation;(b)XRD patterns of ZTN/ZN3 before and after cyclic reactions
為了驗證明異質結光催化劑的對有機染料普適性,測試了ZTN/ZN3對MB、MO和SA的降解實驗(圖7(a~c))。 紫外光照射 60 min 后,ZTN/ZN3 對 MB、MO和SA的降解率分別為87.67%,91.29%和58.55%。其表觀速率常數分別為0.032 27、0.037 31和0.018 1 min-1(圖7d)。結果表明Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑對污染物的降解是非光敏化作用,具有普適性和較強的礦化能力。
2.5 光電化學性能分析
瞬態光電流的大小可以直接反映光催化劑光生電子-空穴對的分離和遷移情況。圖8是不同樣品在紫外光照射下數個開關燈循環間的瞬態光電流響應曲線。光照瞬間樣品的光電流增大,暗光瞬間其光電流又減小為0,說明樣品均具有快速穩定的光電流響應。樣品的光電流大小與光催化活性基本相符。ZTN/ZN3具有最大的瞬態光電流值(5.090 μA),分別為 ZTN1(4.483 μA)和 ZTN2(3.407 μA)瞬態光電流的1.135倍和1.49倍。說明Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑與Zn2SnO4-xNx相比具有更高的光生電子-空穴對的分離效率。ZTN/ZN1、ZTN/ZN2、ZTN/ZN4 和ZTN/ZN5的光電流值明顯低于ZTN1和ZTN2,源于ZnO1-yNy的光腐蝕特性。此外,當N摻雜量較小時,表現出基體的光電特性;當N雜質元素的含量過高時,成為光生電子-空穴對的復合中心,抑制光生載流子的分離,因而ZTN/ZN4和ZTN/ZN5的光電流較小。ZTN/ZN3最大的光電流值源于其適中的N摻雜量和ZnO1-yNy含量有利于構筑界面異質結和雙雜質能級效應,提高了光催化活性。
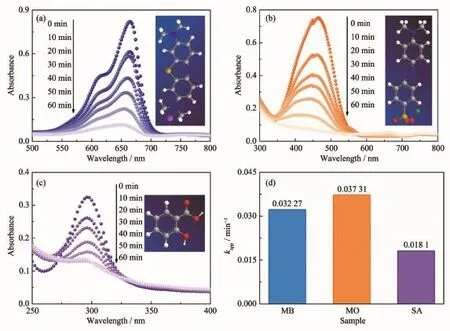
圖7 ZTN/ZN3在紫外光照射下對(a)MB、(b)MO和(c)SA的降解曲線及(d)表觀速率常數Fig.7 Degradation curves of(a)MB,(b)MO and(c)SA over ZTN/ZN3 under UV light irradiation and(d)corresponding apparent rate constants
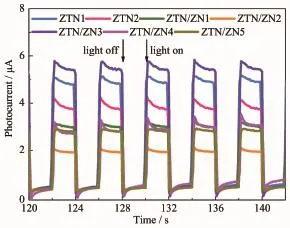
圖8 樣品在紫外光照射下的瞬態光電流響應Fig.8 Transient photocurrent response of samples under UV light irradiation
圖9 是不同樣品在紫外光照射下的電化學阻抗圖譜(EIS)。曲線的圓弧半徑越小,樣品光生電子-空穴對的遷移阻力越小,載流子在界面的遷移速率越快[31]。EIS測試結果與瞬態光電流測試結果基本相符。ZTN/ZN3的阻抗圖具有最小的圓弧半徑(圖9),即ZTN/ZN3具有最小的光生載流子遷移阻力。使用ZSimpWin軟件對樣品的Nyquist點圖進行擬合,所選擬合電路為 Rs(QRct),其中 Rs、Q、Rct分別代表溶液電阻、常相位角元件和電荷轉移電阻。擬合結果如表1所示。表中n值與Q元件的特性有關(n=1,代表理想電容;n=0,代表純電阻;n=-1,代表電感;n=1/2,代表Warburg擴散阻抗;0<n<1,則為常相位角原件Q)。ZTN1、ZTN2和ZTN/ZN3的電荷轉移電阻分別為75.89、97.57和 40.07 kΩ,ZTN/ZN3號樣品的電荷轉移電阻最小,表明Zn2SnO4-xNx和ZnO1-yNy間界面異質結的形成可以減小光生載流子的遷移阻力,加快界面電荷轉移,從而提高光生電子-空穴對的分離效率,達到改善其光催化性能的目的。
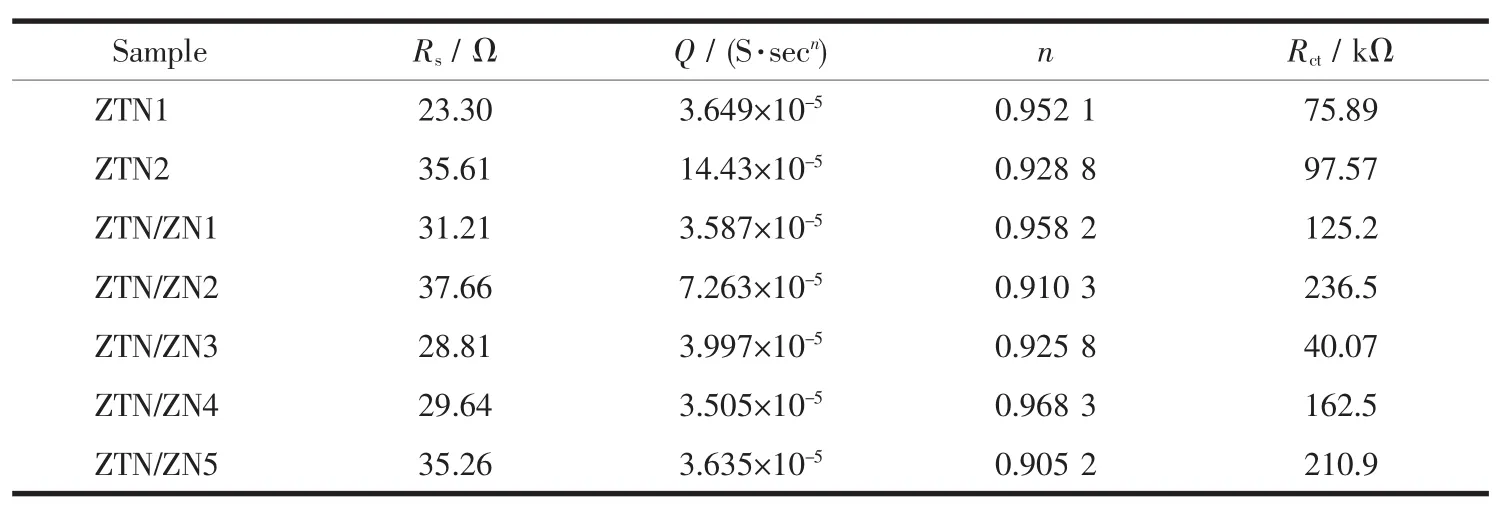
表1 所有樣品Nyquist點圖的擬合結果Table 1 Fitting results of Nyquist plots of all samples
2.6 Zn2SnO4-x N x/ZnO1-y N y的光催化反應機理分析
為了進一步分析Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑的光催化反應機理,并了解各種活性物種在光催化反應過程中所起的作用,以ZTN/ZN3作為光催化劑,向光催化系統中分別加入對苯醌(BQ,0.001 g)、乙二胺四乙酸二鈉(EDTA-2Na,0.37 g)和叔丁醇(tBuOH,1 mL)作為超氧基(·O2-)、空穴(h+)和羥基自由基(·OH)的捕獲劑,進行活性物種捕獲實驗。紫外光照射40 min后,加入3種捕獲劑后對RhB的降解率由95.19%分別減小到34.00%、41.45%和50.37%,說明·O2-、h+和·OH 均為光催化反應期間起主要作用的活性物種。
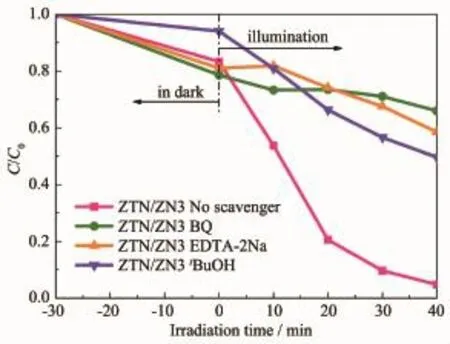
圖10 不同捕獲基對Zn2SnO4-x N x/ZnO1-y N y光降解RhB的影響Fig.10 Effect of different trapping agents on the photodegradation of RhB over Zn2SnO4-x N x/ZnO1-y N y photocatalyst
半導體的禁帶寬度可以通過Kubelka-Munk公式(10)估算:

式中,α、h、ν、Eg分別代表樣品的光吸收系數、 入射光頻率、普朗克常數和禁帶寬度。A為常數,n與半導體類型有關。Zn2SnO4-xNx和ZnO1-yNy均為直接帶隙半導體,其n值為1[32-33]。根據公式(10),計算得到Zn2SnO4-xNx(ZTN1)和ZnO1-yNy(采用同樣的方法制備得到的純相ZnO1-yNy)的禁帶寬度分別為3.82和3.19 eV。 圖11b為Zn2SnO4-xNx和ZnO1-yNy的Mott-Schottky點圖,正的斜率表明Zn2SnO4-xNx和ZnO1-yNy均為n型半導體。對于n型半導體而言導帶電勢約等于平帶電勢。根據Mott-Schottky點圖中的直線部分在x軸上的截距可知Zn2SnO4-xNx和ZnO1-yNy的導帶電勢約為-0.73和-0.25 eV。根據公式(11):

式中,EV、Eg和EC分別為半導體的價帶電勢、禁帶寬度和導帶電勢,計算得到Zn2SnO4-xNx和ZnO1-yNy的價帶電勢分別為3.09和2.94 eV。
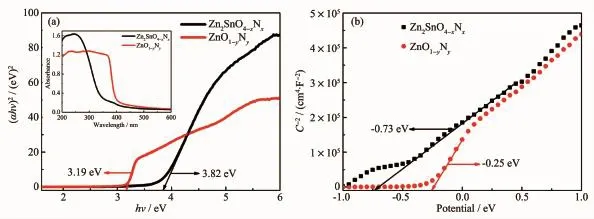
圖 11 Zn2SnO4-x N x和 ZnO1-y N y的(a)(αhν)2-hν圖和(b)Mott-Schottky 點圖Fig.11 (a)Plots of the(αhν)2 vs hν and(b)Mott-Schottky plots of Zn2SnO4-x N x and ZnO1-y N y
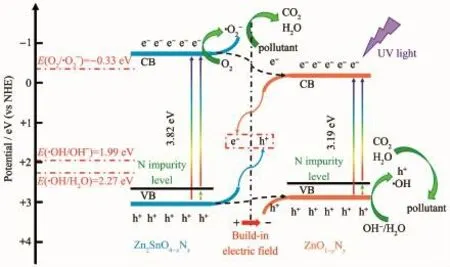
圖12 Zn2SnO4-x N x/ZnO1-y N y異質結光催化劑的光催化機理圖Fig.12 Photocatalytic mechanism of Zn2SnO4-x N x/ZnO1-y N y heterojunction photocatalyst
圖12 是Zn2SnO4-xNx/ZnO1-yNy異質結的光催化機理圖。在紫外光照射下,寬禁帶的Zn2SnO4-xNx和ZnO1-yNy均可以被激發,價帶電子吸收光子能量,躍遷到導帶位置,同時在價帶留下空穴。同時,N雜質能級可以作為電子躍遷的跳板,光生電子可以先躍遷到N雜質能級,再由雜質能級躍遷至導帶位置,兩步躍遷過程可以有效提高半導體的光響應范圍和光生載流子的分離效率;此外,由于N原子是一個良好的電子供體,其電荷可直接被激發到導帶位置參與光催化反應。根據傳統的異質結理論,由于Zn2SnO4-xNx(-0.73 eV)的導帶電勢比ZnO1-yNy(-0.25 eV)的導帶電勢更負,價帶電勢(Zn2SnO4-xNx:3.09 eV)比ZnO1-yNy(2.94 eV)的價帶電勢更正,Zn2SnO4-xNx的光生電子和空穴傾向于遷移到ZnO1-yNy的導帶和價帶位置,如圖12黑色虛線箭頭所示,形成傳統的Ⅰ型異質結。根據活性物種捕獲實驗結果,·O2-物種在光催化降解RhB過程中起到了很關鍵的作用。但是,在這種情況下,由于·O2-/O2(-0.33 eV vs NHE)[34]的電勢比ZnO1-yNy的導帶電勢更負,氧氣分子無法與ZnO1-yNy的導帶電子反應生成·O2-物種,與活性物種捕獲實驗結果不符。基于以上分析,Zn2SnO4-xNx/ZnO1-yNy復合光催化劑并不是遵循傳統的Ⅰ型異質結。根據之前的報道[35-36],Zn2SnO4和ZnO的功函分別為4.81和5.10 eV,Zn2SnO4的電子逸出功較小,因而,Zn2SnO4-xNx的表面電子將會向ZnO1-yNy轉移,導致ZnO1-yNy的表面電荷密度增大,而Zn2SnO4-xNx的表面電荷密度減小,由此在界面處形成一個由Zn2SnO4-xNx指向ZnO1-yNy的內建電場,如圖12紅色箭頭所示,并且這一電荷轉移過程使Zn2SnO4-xNx的能帶向上彎曲,ZnO1-yNy的能帶向下彎曲,在兩者的界面處,Ⅰ型異質結轉變為Ⅱ型異質結。在內建電場作用下,ZnO1-yNy的光生電子很容易與Zn2SnO4-xNx的光生空穴復合,如圖12中金黃色和藍色實線箭頭所示,Ⅱ型異質結再進一步轉變為Z型異質結,留下具有更高還原電勢的光生電子參與之后的反應。Zn2SnO4-xNx的導帶電子將與氧氣分子反應生成·O2-物種,而ZnO1-yNy的價帶空穴則與水分子或者OH-反應生成·OH 物種。在·O2-、·OH和h+三者的共同作用下將有機污染物降解成為水和二氧化碳或者其他無毒性的小分子。因此,Zn2SnO4-xNx/ZnO1-yNy增強的光催化活性可歸因于雙雜質能級和Z型異質結的協同作用。
3 結 論
通過控制反應前驅液中水合肼的濃度,采用微波溶劑熱法一步合成具有不同ZnO含量的核殼結構Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑,其在紫外光照射下對有機污染物的降解具有較強的礦化能力、普適性、良好的循環穩定性和非光敏化作用。由于不同功函所導致的界面電荷轉移和能帶彎曲使兩者由傳統的Ⅰ型異質結轉變為Ⅱ型異質結,再由界面處內建電場的作用使其由Ⅱ型異質結轉變為Z型異質結。N原子進入Zn2SnO4和ZnO的晶格中形成雙雜質能級,拓寬了其光響應范圍;Zn2SnO4-xNx顆粒附著在ZnO1-yNy納米棒表面形成的核殼結構可緩解ZnO1-yNy的光腐蝕現象。Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑降解RhB的速率和瞬態光電流值分別為純相Zn2SnO4-xNx的1.40~1.43倍和1.14~1.49倍,光催化性能的提高主要歸因于Z型異質結和雙雜質能級的協同作用。這種新型的直接Z型Zn2SnO4-xNx/ZnO1-yNy異質結光催化劑在環境凈化領域具有潛在的應用前景。
