B摻雜的石墨烯作為鈉離子電池負極材料的研究
2019-07-08 09:09:28姚利花甄海龍
原子與分子物理學報 2019年3期
關鍵詞:結構
姚利花, 甄海龍
(1. 山西大同大學機電工程學院,大同 037003; 2. 山西大同大學物理與電子科學學院, 大同 037009)
1 引 言
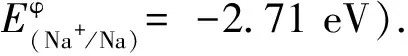
石墨烯是具有單原子尺寸厚度的六角蜂窩狀結構材料.近年來,石墨烯由于其奇異性能引起了高度重視,其高的儲存能力使它有望成為二次電池的電極材料.許多結果表明石墨烯具有高的儲存能力[8-11],Yoo等[10]發現鋰在石墨烯的儲存量達到784 mAh/g,是鋰在石墨中儲存量的2倍.原因是石墨烯的二維六角蜂窩結構為鋰原子的儲存提供了額外的位點.因此,石墨烯有望成為鈉離子電池的電極材料.而且,硼摻雜的碳材料比相應的純碳材料表現出更高的可逆容量和循環穩定性[12,13].
第一性原理是一種研究和預測材料性能的重要方法,近年來得到廣泛的應用.例如,2017年,姚等[14]利用第一性原理研究了B、N摻雜對鈉在石墨烯上吸附性能的影響.余等[15]利用第一性原理研究了壓力下CaN2的結構穩定性和電子結構.2018年,歐等[16]利用第一性原理研究了單層MoS2表面吸附Ag6團簇電子結構;陽等[17]利用第一性原理研究了二氧化鈰基稀磁氧化物的磁性.
基于鈉的上述優點,鈉離子電池有望成為新型二次電池,但目前研究表明吸附在石墨烯上的鈉容易產生枝晶或團簇[18].因此,本文采用第一性原理對硼摻雜的石墨烯作為鈉離子電池電極材料進行相關研究.主要研究鈉吸附在石墨烯上的吸附能、電荷密度、態密度和儲鈉量.
2 模擬方法和計算模型
2.1 模擬方法
采用基于密度泛函理論(DFT)的第一性原理計算方法,并使用Materials Studio軟件中的DMol3程序包.電子交換關聯勢采用廣義梯度近似(GGA)下的PW91泛函進行處理.與LDA相比,GGA不會導致分子間的強烈結合[19,20]. 計算中取X和Y方向在石墨烯平面內,Z方向垂直于石墨烯平面,真空層取20 ?.在倒易的k空間中,軌道截止選取5.1 ?.自洽迭代過程簡約布里淵區積分k點使用Monkhorst-Pack方法[21]選取2×2×1格點,計算中自旋不受限制,并且選擇標準自旋為初始自旋,在做自洽計算時總能量誤差不大于1.0×10-6hartree.
2.2 計算模型
如圖1所示,是三種石墨烯的計算模型,它是由4×4×1個六角碳環(共32個碳原子)組成,碳原子以六方形的蜂窩狀點陣有序排列在二維平面上.在P-graphene上,主要考慮三個高對稱吸附位置:T位于C原子的正上方;B位于C—C鍵中點的上方;H位于六方蜂巢格子中心的正上方.在Defect-Ⅰ上,主要考慮四個高對稱吸附位置:TC位于碳原子的正上方;TB位于硼原子的正上方;B位于C—B鍵中點的上方;H位于六方蜂巢格子中心的正上方.在Defect-Ⅱ上,主要考慮三個高對稱吸附位置:T位于硼原子的正上方;B位于C—B鍵中點的上方;H位于六方蜂巢格子中心的正上方.

(1)
這里,F是法拉第常數.吉布斯自由能ΔG的變化可以表示為:
ΔG=ΔE+PΔV-TΔS
(2)
其中,PΔV和TΔS分別為反應前后的體積變化和熵值變化而導致的吉布斯自由能變化項,由于通常情況下這兩項遠小于ΔE項,因此:
ΔG≈ΔE
(3)
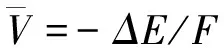
(4)
而ΔE為吸附鈉前后石墨烯體系的能量之差,對應于吸附能Ead:
Ead=[EGraphene+Na-(nENa+EGraphene)]/n
(5)
式中,EGraphene+Na是吸附鈉后石墨烯的總能量,ENa是鈉原子的總能量,EGraphene是吸附鈉前石墨烯的總能量,n是鈉原子的個數.
3 結果與討論
3.1 吸附位置的確定
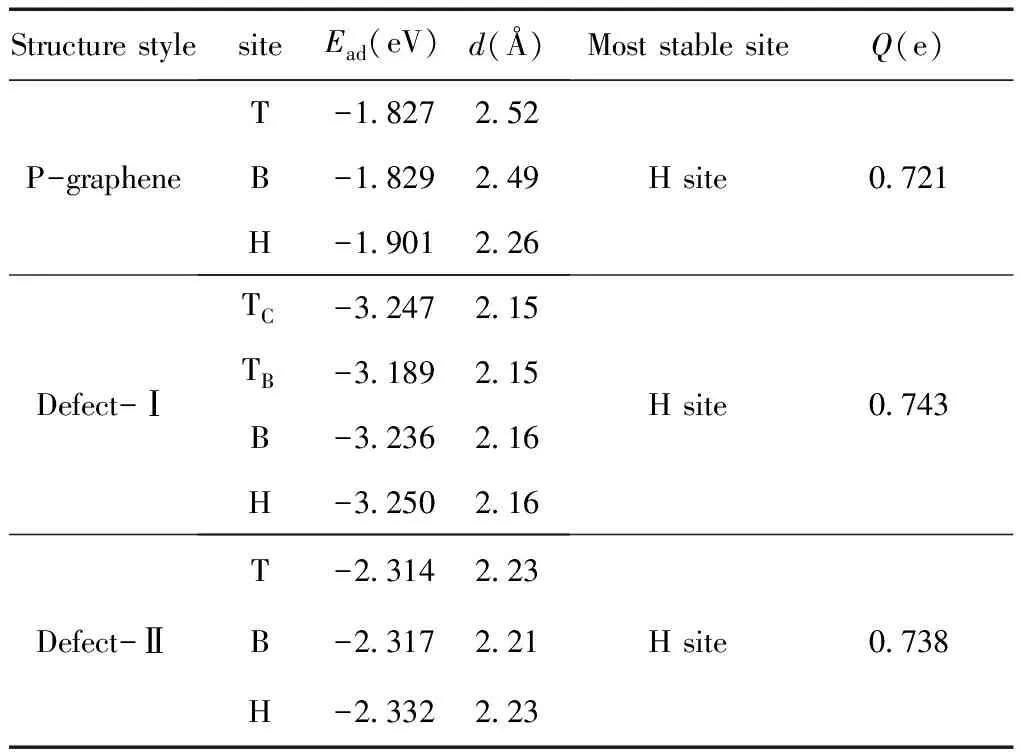
如圖1所示,在P-graphene、Defect-Ⅰ和Defect-Ⅱ中,把鈉原子放在高對稱位上,使其與石墨烯平面的垂直距離相等進行結構優化.表1列出了優化至最穩定結構時鈉原子在各個吸附位置的吸附能和鈉原子與石墨烯平面的垂直距離.可以看出,三種石墨烯中,鈉原子在H位上吸附時吸附能最低,且鈉原子與石墨烯的垂直距離較小.這是由于對于一個離子鍵的金屬原子來說,它的平衡高度取決于金屬原子和石墨烯相反電荷的吸引以及短程電子的排斥作用[25],由于H位的電荷密度低于B位和T位(圖2(a)、(b)和(c)),金屬原子更加容易趨于穩定.因此,H位是最穩定的吸附位置.

圖1 (a)、(b)和(c)分別是P-graphene,Defect-Ⅰ和Defect-Ⅱ的計算模型.灰色球代表碳原子,粉色球代表硼原子,T、TC、TB、B、H代表吸附位置.Fig. 1 The calculation models of P-graphene (a),Defect-Ⅰ (b) and Defect-Ⅱ (c). Grey spheres represent carbon atoms, pink spheres represent boron atoms. T, TC, TB, B and H represent the adsorption sites.
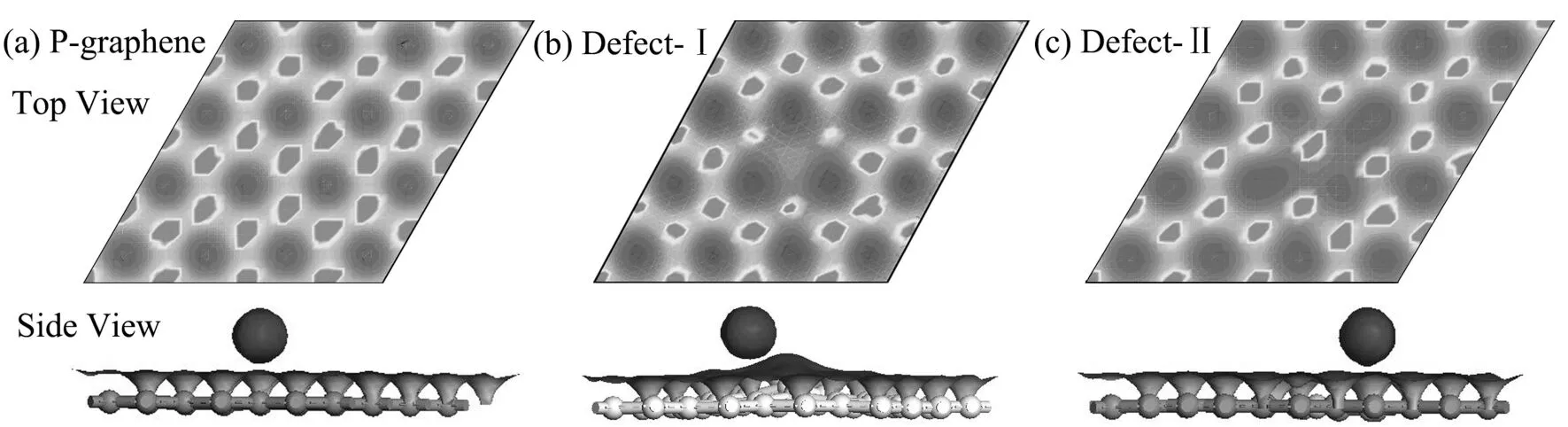
圖2 (a)、(b)和(c)分別是P-graphene,Defect-Ⅰ和Defect-Ⅱ的電荷密度.Fig. 2 The charge densities of P-graphene (a),Defect-Ⅰ (b) and Defect-Ⅱ (c).
3.2 電荷密度
從表1可以看出,鈉原子吸附在P-graphene、Defect-Ⅰ和Defect-Ⅱ上H位的吸附能分別是-1.901 eV,-3.250 eV和-2.332 eV;鈉原子與三種石墨烯的垂直距離分別為2.26 ?,2.16 ?和2.23 ?;傳輸電荷分別為0.721e,0.743e和0.738e.這說明與P-graphene相比,鈉原子在Defect-Ⅰ和Defect-Ⅱ上吸附的吸附能和傳輸電荷增大,垂直距離減小.因此,鈉原子與Defect-Ⅰ和Defect-Ⅱ之間的相互作用增強,硼原子的引入使得鈉原子在石墨烯上吸附更容易.這可以從電荷密度來解釋,如圖2(b)和(c) Top view所示,Defect-Ⅰ和Defect-Ⅱ中H處的電荷密度較低,提供了空穴,圖2(b)和(c) Side view所示,Defect-Ⅰ和Defect-Ⅱ與鈉原子的距離較小,電荷從鈉原子傳輸到石墨烯更容易,相互作用更強,這與Mpourmpakis等[26]的報道相符合.因此,硼摻雜的石墨烯更適合儲鈉.這與Wang等[27]關于摻雜硼原子使得鋰在石墨烯上吸附能力提高的結果相一致.
3.3 態密度
為了研究石墨烯H位對鈉原子的吸附機制,計算了鈉-石墨烯體系的分波態密度.圖3為吸附鈉原子后石墨烯的分波態密度.從圖中可以看出,鈉原子只對導帶有貢獻,這說明鈉是完全離子化的,且其與石墨烯的相互作用力主要是庫侖力[28].從圖3看出,P-graphene中沒有發生軌道雜化現象,Defect-Ⅰ和Defect-Ⅱ中,分別在費米能級以上3.791 eV和5.17 eV處,硼原子的p軌道與鈉原子的p軌道發生雜化,這是共價鍵的特征,很好的解釋了Defect-Ⅰ和Defect-Ⅱ對鈉原子強的吸附作用,P-graphene對鈉原子的吸附作用較弱[29].
表1 石墨烯上鈉原子處于不同吸附位置時的吸附能(Ead),Na原子與石墨烯平面的垂直距離(d)和鈉原子處于最穩定吸附位置時的傳輸電荷.
Table 1 The adsorption energies (Ead) and the Na atom-graphene sheet distances(d) of different adsorption sites, and the charge transfers (Q) of most stable site from the Na atom to graphenes.
Structure stylesiteEad(eV)d(?)Most stable siteQ(e) P-grapheneT-1.8272.52B-1.8292.49H-1.9012.26H site0.721Defect-ⅠTC-3.2472.15TB-3.1892.15B-3.2362.16H-3.2502.16H site0.743Defect-ⅡT-2.3142.23B-2.3172.21H-2.3322.23H site0.738
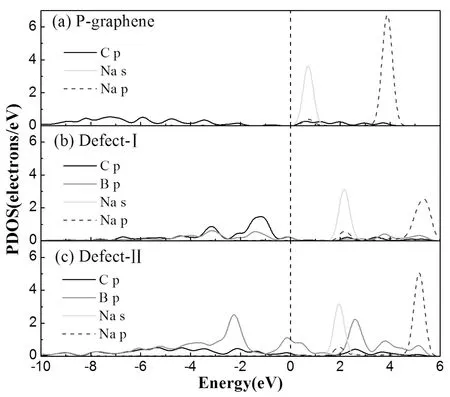
圖3 P-graphene,Defect-Ⅰ和Defect-Ⅱ吸附鈉原子后的分波態密度.Fig. 3 The partial densities of states (PDOSs) of P-graphene (a),Defect-Ⅰ (b) and Defect-Ⅱ (c) after Na adsorption, the dashed line at zero indicates the Fermi level.
3.4 吸附量
對于鈉離子電池電極材料來說,儲鈉量是一個非常關鍵的因素.將鈉原子分別放在與石墨烯平面的垂直距離相等的位置上進行結構優化,圖4是鈉原子的平均吸附能與鈉原子個數之間的關系,可以看出,當P-graphene、Defect-Ⅰ和Defect-Ⅱ上吸附的鈉原子個數分別為8、10和9時,吸附能分別是-1.104 eV、-0.885 eV和-0.403 eV,都大于鈉原子之間的結合能-1.11 eV[30],說明鈉原子會趨于團聚.

圖4 石墨烯對鈉原子的平均吸附能與鈉原子個數的關系.Fig. 4 The relationship between the average adsorption energies of Na and the number of Na on graphenes.
圖5 (a),(b)和(c)分別是將10個鈉原子放在P-graphene、Defect-Ⅰ和Defect-Ⅱ上優化后的結構.可以看出,結構優化后,與三種石墨烯距離為3.883 ?的鈉原子分別有7、9和8個,且鈉原子分布散亂,但Defect-Ⅱ曲線不光滑,說明在吸附過程中會發生相變[31]. 表2列出了三種石墨烯分別吸附7、9和8個鈉原子時,鈉原子之間的平均距離(dNa-Na),鈉原子在石墨烯上的平均吸附能(Eaad).三種石墨烯中,鈉原子之間的平均距離是分別為-3.237 ?、-3.464 ?和-3.555 ?,遠遠大于Na-Na鍵長1.91 ?[30]; 鈉原子的平均吸附能分別是-1.197 eV、-1.659 eV和-1.246 eV,小于鈉原子的結合能-1.11 eV[30],說明鈉原子不會趨于團聚.因此,P-graphene對鈉原子的吸附量是7,Defect-Ⅰ對鈉原子的吸附量是9,Defect-Ⅱ對鈉原子的吸附量是8,并且都不會趨于團聚.
表2 鈉原子在石墨烯的吸附量,鈉原子之間的平均距離(dNa-Na),鈉原子在石墨烯上的平均吸附能(Eaad),Na-Na鍵長(Lbond)和Na原子的結合能(Ebinding).
Table 2 The storage capacities, the average distances between Na atoms (dNa-Na), the average adsorption energies of Na atoms on graphenes (Eaad), the bond length of the Na-Na bond (Lbond) and the Na-Na binding energy (Ebinding).

Structure styleCapacitydNa-Na(?)Lbond(?)Eaad(eV)Ebinding(eV)P-graphene73.237Defect-Ⅰ93.464Defect-Ⅱ83.5551.91[30]-1.197-1.659-1.246-1.11[30]
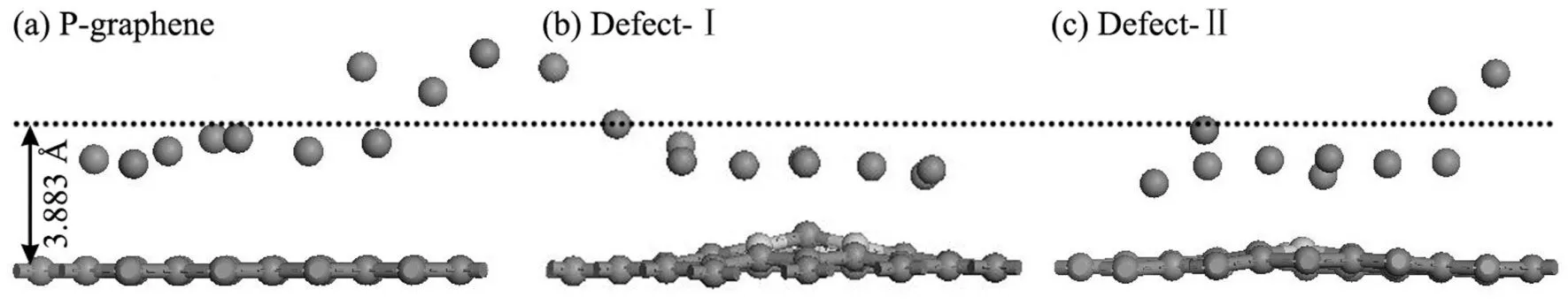
圖5 鈉原子在P-graphene,Defect-Ⅰ和Defect-Ⅱ的吸附結構.灰色球代表碳原子,紫色球代表鈉原子,粉色求代表硼原子.Fig. 5 The adsorption configurations of Na atoms adsorbed on P-graphene (a),Defect-Ⅰ (b) and Defect-Ⅱ (c). Grey spheres represent carbon atoms, purple spheres represent sodium atoms, pink spheres represent boron atoms.
4 結 論
本文研究了P-graphene、Defect-Ⅰ和Defect-Ⅱ三種石墨烯對鈉原子的吸附行為.三種石墨烯中H位是最穩定的吸附位置.與P-graphene相比,Defect-Ⅰ和Defect-Ⅱ對鈉原子的吸附能降低,吸附量提高.P-graphene中不存在軌道雜化現象,而Defect-Ⅰ和Defect-Ⅱ中硼原子與鈉原子發生軌道雜化,說明鈉原子與其相互作用較大.三種石墨烯對鈉原子的吸附量分別是7、9和8個,Defect-Ⅱ吸附鈉原子過程中會發生相變.因此,Defect-Ⅰ更適合儲鈉.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
