石油樣品中金剛烷類化合物的定量分析新方法
2019-06-27 07:11:32王匯彤張水昌魏彩云張朝軍
石油實驗地質 2019年3期
王匯彤,翁 娜,張水昌,魏彩云,張朝軍
(1.中國石油勘探開發研究院,北京 100083;2.提高石油采收率國家重點實驗室,北京 100083)
金剛烷類化合物是原油中一種特殊的環狀烴類化合物,其穩定特性決定了它在地質演變過程中具有很強的抗熱和抗生物降解能力[1-3]。在高成熟原油和凝析油中,甾烷、藿烷等常用生物標記化合物缺失,金剛烷類化合物可成為判定成熟度的重要參數[4-6],也可用于研究油氣運移方向、油源判識和判別原油裂解程度[7-9]。
如何得到金剛烷系列化合物的絕對含量一直是地球化學家們嘗試解決的難題。由于金剛烷類化合物在石油地質樣品中的含量較低,受共餾峰干擾和提純條件的限制,無法用常規氣相色譜(GC-FID)對其進行絕對定量分析。目前常用的定量方法是氣相色譜—質譜(GC-MS)的內標半定量法。在質譜分析中,嚴格的絕對定量應該采用與目標化合物結構相同、具有相同特征離子特征或碎裂方式的化合物作為內標化合物,才能得到較為準確的定量結果。在國外,WEI等[3,10]使用6種不同結構的氘代金剛烷化合物作為內標物質,得到的定量結果較可靠。在國內,由于標樣的缺乏,通常只用一種結構的氘代金剛烷來定量所有結構的金剛烷系列化合物,得到的結果與實際偏差較大。馬安來等[11]用氘代單金剛烷做內標,確定出雙金剛烷、氘代甲基雙金剛烷和氘代二甲基單金剛烷在GC-MS上的響應因子,并以此計算出雙金剛烷、甲基—單金剛烷、二甲基—單金剛烷的絕對含量,用于判別塔河油田的原油裂解程度。但對于其他結構的金剛烷系列化合物,目前沒有有效的絕對定量方法,這就限制了金剛烷類化合物在地球化學上的研究工作的開展。
全二維氣相色譜(GC×GC)是一種分離復雜混合物的新技術,它的二維正交色譜柱系統使得一些在常規氣相色譜上因沸點相同而共餾的化合物,在第二維色譜柱上可以根據極性被很好地分開。聯用的FID檢測器對所有碳氫結構化合物的響應因子近似為1,因此GC×GC-FID被認為是目前定量碳氫結構化合物的最有效方法之一[12-13]。本文在早期工作基礎上[14-16],建立了一套完整的石油地質樣品中金剛烷系列化合物的色譜定量方法,為金剛烷類化合物的地球化學研究提供有效的技術支持。
1 實驗部分
1.1 儀器與設備
全二維氣相色譜—飛行時間質譜儀(美國LECO公司的Pegasus 4D),工作站為Chroma TOF軟件,氫火焰離子化檢測器(美國Agilent公司),Trace氣相色譜/ DSQⅡ質譜儀(美國Thermo Fisher公司),氮吹儀(美國進口)。
1.2 試劑與材料
分析純的正己烷、二氯甲烷、三氯甲烷均購自國藥集團化學試劑有限公司,使用前進一步提純;細硅膠(100~200 目,200 ℃下活化4 h)。
1.3 樣品
本文共選取2個巖石抽提物和7個原油樣品進行分析(表1)。
1.4 實驗條件
1.4.1 GC×GC-TOFMS分析條件
一維色譜柱是30 m×0.25 mm×0.25 μm 的DB1-MS柱(美國Agilent公司),升溫程序為50 ℃保持0.2 min,以2 ℃/min的速率升到200 ℃保持0.2 min,再以8 ℃/min的速率升到300 ℃保持10 min。二維色譜柱是1.5 m×0.1 mm×0.1 μm 的DB-17HT柱(美國Agilent公司),采用與一維色譜相同的升溫速率,溫度比一維色譜高5 ℃。調制器溫度比一維色譜高45 ℃。以He氣為載氣,流速設定為1 mL/min。調制周期10 s,其中2.5 s熱吹時間。進樣口溫度為300 ℃,分流進樣模式。對于凝析油樣品,分流比700∶1,進樣量0.5 μL。對于其他原油或者巖石抽提物樣品,分流比為20∶1,進樣量為1 μL。
飛行時間質譜的傳輸線和離子源溫度分別為300 ℃和240 ℃,檢測器電壓1 500 V,質量掃描范圍40~520 amu,采集速率100 譜圖/s。對于凝析油樣品,溶劑延遲時間為0 min;對于其他原油或者巖石抽提物樣品,溶劑延遲時間為10 min。
1.4.2 GC×GC-FID分析條件
采用與GC×GC-TOFMS相同的色譜實驗條件。FID檢測器中載氣、氫氣、空氣的流速分別為23,30,400 mL/min。檢測器溫度310 ℃,采集頻率200 譜圖/s,溶劑延遲時間與TOFMS設置一致。
1.4.3 GC-MS分析條件
色譜柱為30 m×0.25 mm×0.25 μm 的DB1-MS柱,氦氣為載氣,流速為1 mL/min。升溫程序為:起始溫度50 ℃,以15 ℃/min的速率升到80 ℃,再以2 ℃/min的速率升到230 ℃,再以25 ℃/min的速率升到310 ℃保持20 min。進樣口溫度為280 ℃,不分流進樣模式,進樣量1 μL。質譜檢測器電壓1600V,選擇離子方式掃描,溶劑延遲時間為10 min。
1.5 定量方法
1.5.1 GC×GC-FID定量方法
利用GC×GC-TOFMS和GC×GC-FID分別分析待測樣品,得到外觀一致的GC×GC-TOFMS在總離子流(TIC)下的色譜圖和GC×GC-FID的色譜圖。結合文獻[17]和TOFMS提供的質譜信息,在GC×GC-TOFMS圖譜上定性識別出標樣和各金剛烷類化合物。根據化合物在GC×GC-TOFMS上相對保留時間,在GC×GC-FID色譜圖上標記相應的目標化合物,并得到其峰面積,采用內標法計算得到定量結果。
1.5.2 GC-MS定量方法
利用GC-MS的選擇離子模式分析待測樣品,得到樣品的色譜—質譜總離子流譜圖和在選擇離子m/z135、136、149、152、163、177、187、188、201、215、239、240、253、267下的色譜—質譜圖。手動積分得到不同選擇離子下目標化合物的峰面積,利用內標法計算得到其定量結果。
2 結果與討論
2.1 分析條件的選擇
2.1.1 前處理方法的選擇
金剛烷類化合物相對含量較高的凝析油樣品,可以不進行任何前處理,直接用GC×GC-FID分析。取適量凝析油樣品于1.5 mL自動進樣瓶中,稱重后加入配制好的D16-單金剛烷標準溶液(溶劑為CH2Cl2)和適量CH2Cl2溶劑,直接進樣分析即可。該方法避免了前處理過程中低沸點金剛烷的損失,保證了定量結果的準確性。圖1a為S9號凝析油樣品直接進樣分析的GC×GC-FID譜圖,從圖上可看出,17個單金剛烷化合物在GC×GC-FID下能夠得到很好的分離。本實驗選擇S9樣品為標準樣品,用來確定其他樣品中金剛烷類化合物在GC×GC-FID上的出峰位置。
對于正常原油或者稠油樣品,金剛烷類化合物相對含量較低,易受單環芳烴的干擾而不易被檢出(圖1b)。用傳統的石油行業標準中的粗硅膠層析柱法對樣品進行前處理(圖2a),得到的飽和烴餾分中易混入烷基苯和單芳甾[18],影響金剛烷類化合物的GC×GC-FID定量分析。王匯彤等[19]改進了分離方法(圖2b),得到的飽和烴餾分中不混有烷基苯,可用于金剛烷類化合物的定量。本文在該方法的基礎上進行改進,采用小柱法,用市售的長滴管作為玻璃層析柱,僅填充1 g細硅膠,取5 mg左右的原油樣品,用1.5 mL正己烷淋洗(圖2c),得到的飽和烴前餾分既可滿足金剛烷類化合物的GC×GC-FID定量要求,又節省了原料和分析時間,同時降低了單金剛烷的揮發損失,特別適合少量樣品的分析需求。
傳統的分析巖石樣品中金剛烷類化合物的方法,是先用CHCl3進行抽提,揮干后再用正己烷溶解,采用圖2a或2b的方法得到其飽和烴組分。但是在揮干CHCl3過程中,C13之前的化合物損失嚴重(圖3b),單金剛烷類化合物無法檢測到。為了避免揮干損失,本文巖石樣品先抽提,CHCl3揮干少于1 mL時,加入少量細硅膠與溶液混合均勻,晾干,在圖2c的小柱下部填充細硅膠,上部填充混有樣品的細硅膠,然后再進行分離,這樣單金剛烷化合物可以被保留(圖3a)。
為驗證該前處理方法的可靠性,用CHCl3配制了濃度分別為0.25 mg/L、0.43 mg/L的單金剛烷和雙金剛烷混合溶液做回收率實驗,得到單金剛烷的回收率為98.2%,雙金剛烷的回收率為99.1%,滿足測定需要。
2.1.2 全二維氣相色譜分析方法的優化
王匯彤等[14]在分析凝析油樣品時,有提及用GC×GC-FID定量金剛烷類化合物。但該方法為滿足nC3—nC8之間化合物的分離,前期升溫速率慢,所用分析時間長。本文對該方法進行改進,選擇較短的色譜柱,降低成本。由于前處理過程中重組分收集少,因此采用前期慢、后期快的升溫速率,節省分析時間。為了與GC-MS的分析結果對比,載氣也采用1 mL/min的流速。
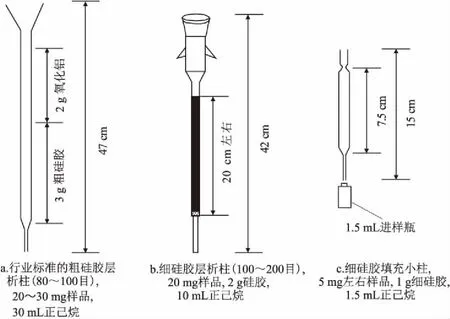
圖2 制備飽和烴組分的分離方法示意
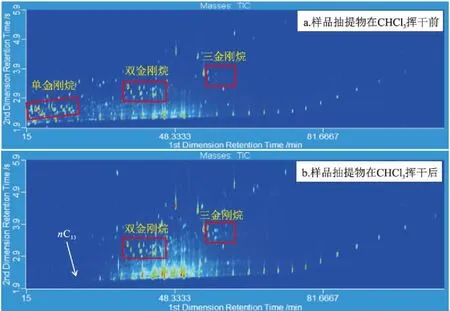
圖3 S2號巖石樣品用GC×GC-FID分析的全二維輪廓圖
2.2 金剛烷類化合物的分析結果
2.2.1 GC×GC-TOFMS定性結果
用GC×GC-TOFMS分析金剛烷類化合物,在特征離子m/z149 下,一維色譜上只有一個色譜峰(圖4a標記的“1”號峰),而二維色譜上有2個色譜峰(圖4a標記的“1-1”和“1-2”號峰),其特征離子均為m/z149,分子離子峰分別為m/z164和m/z178。在已知文獻中,“1”號峰是1-乙基-3-甲基單金剛烷[20],因此“1-2”號峰才是目標化合物。“1-1”和“1-2”號峰在一維色譜上是共餾峰,但在GC×GC上就能被很好地分離。同理在特征離子m/z163下也存在這種情況(圖4b)。由此可見,用GC×GC分析金剛烷類化合物,可以消除共餾峰的影響,無需借助質譜,在GC×GC-FID下就能實現很好分離(圖5)。
2.2.2 GC×GC-FID的定量結果
根據GC×GC-TOFMS的定性信息,在GC×GC-FID譜圖上依照相對保留時間標記出常用的17個單金剛烷、9個雙金剛烷以及內標物質的出峰位置(圖5),利用Chroma TOF軟件得到這些化合物的峰面積積分結果。對于需要前處理的樣品,金剛烷類定量結果與真實值有一定差距,主要反映在前處理過程中化合物的揮發損失上。本實驗采用的小柱分離法,避免了樣品濃縮過程中金剛烷類化合物的揮發損失,結果與真實值更加接近。表2列出了8個樣品中金剛烷的定量結果,其中S1、S2的結果是巖石樣品中的金剛烷含量;S9樣品是標準樣品,未進行前處理,僅用來確定金剛烷在GC×GC-FID上的出峰位置,因此表中未給出S9的定量結果。
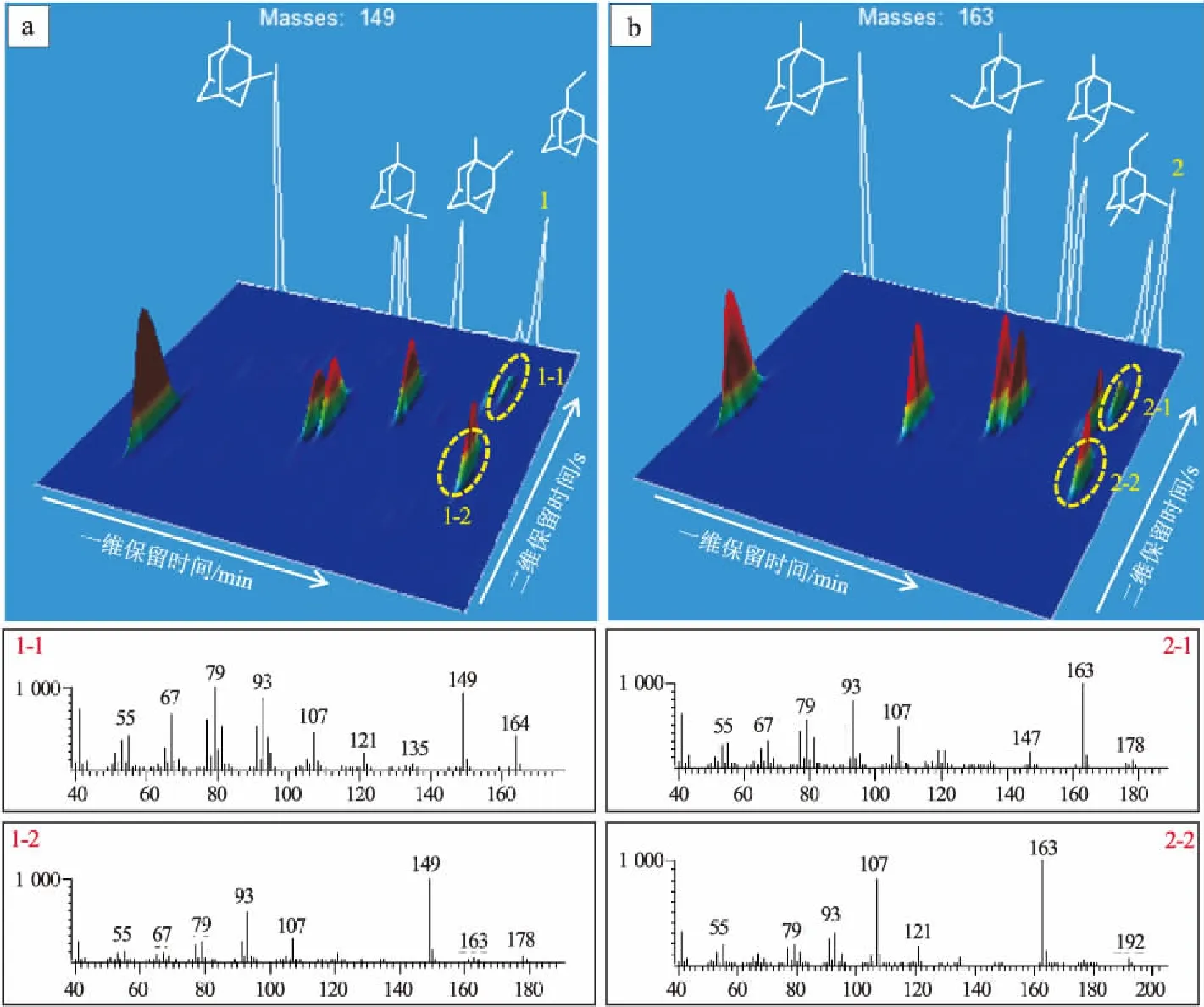
圖4 S9號樣品的GC×GC-TOFMS全二維3D譜圖
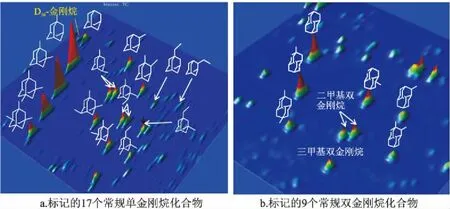
圖5 S9號樣品的GC×GC-FID下全二維3D譜圖
2.3 GC×GC-FID與GC-MS方法分析結果的比較
WANG等[16]已經驗證過GC×GC-FID定量的可靠性,發現其與真實定量結果最接近,誤差小于5% 。而GC-MS的定量結果與真實結果相差很大,原因是GC-MS定量分析時,需要大量、不同結構的標準樣品,但是在實際工作中很難得到。本文用GC-MS與GC×GC-FID對6個正常原油樣品(S3~S8)進行分析(表3),并進行了對比(表4)。
從表4可以看出,24個化合物中只有單金剛烷的定量結果在2臺儀器間的偏差小于5%,原因是本次試驗選用的內標物質是D16-金剛烷,該化合物在結構上與單金剛烷最為接近,因此結果偏差最小。表4中還列出2個常用的金剛烷地化參數間的對比情況[8,20],結果顯示標樣的選取不僅對單體化合物的定量結果有影響,對地化參數也有影響,相比于單金剛烷,雙金剛烷的地化參數影響相對較少。
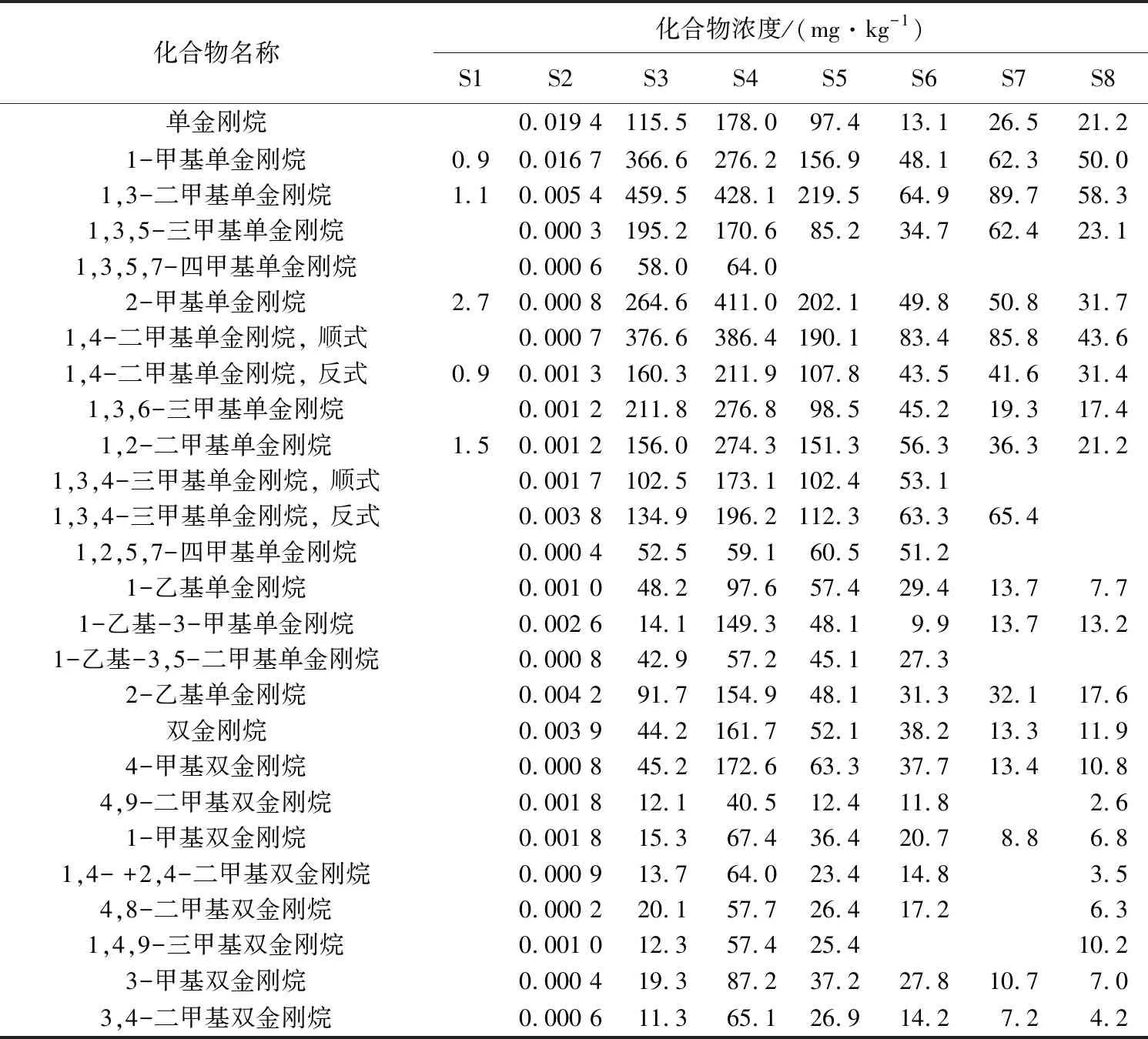
表2 金剛烷類化合物的GC×GC—FID定量結果
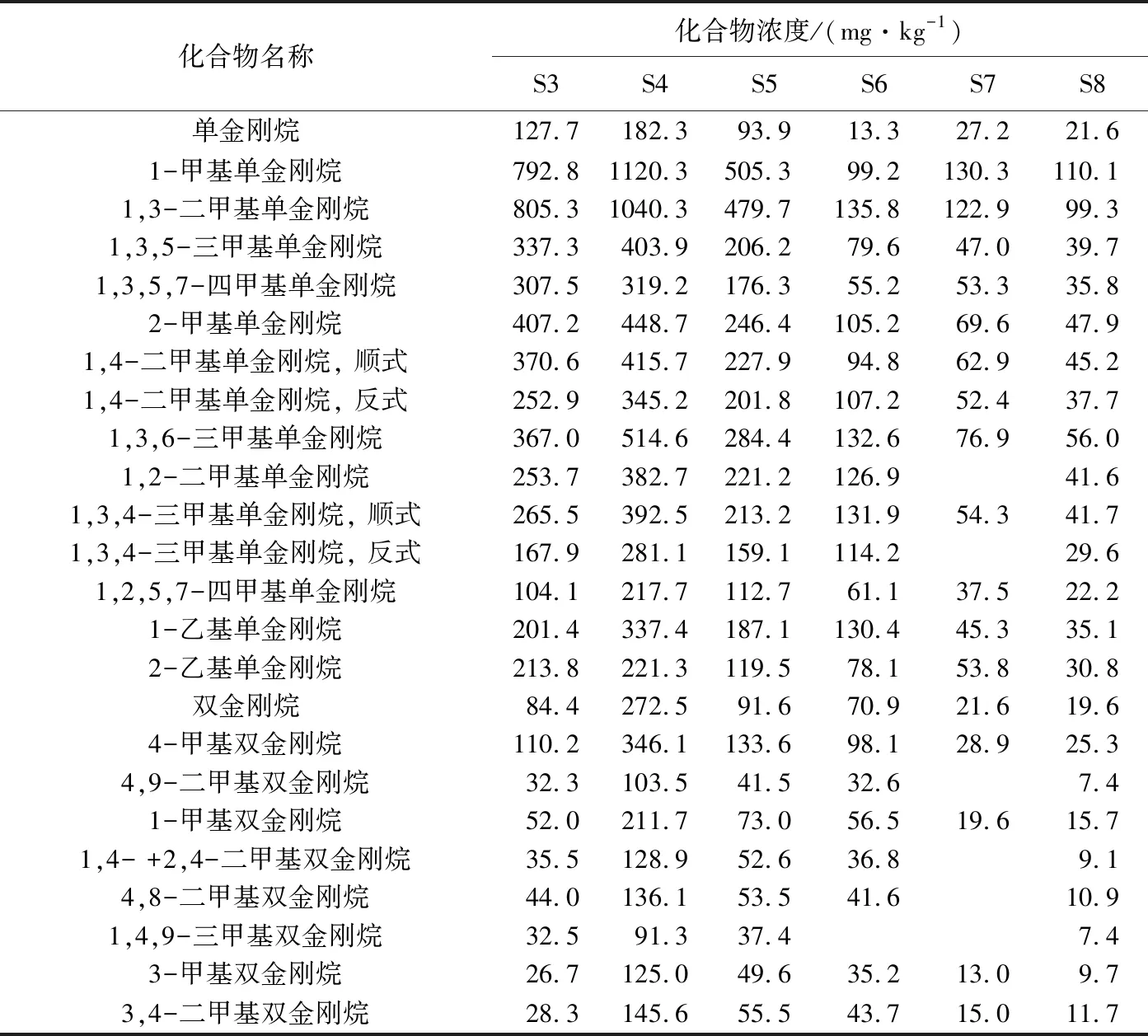
表3 金剛烷類化合物的GC-MS定量結果
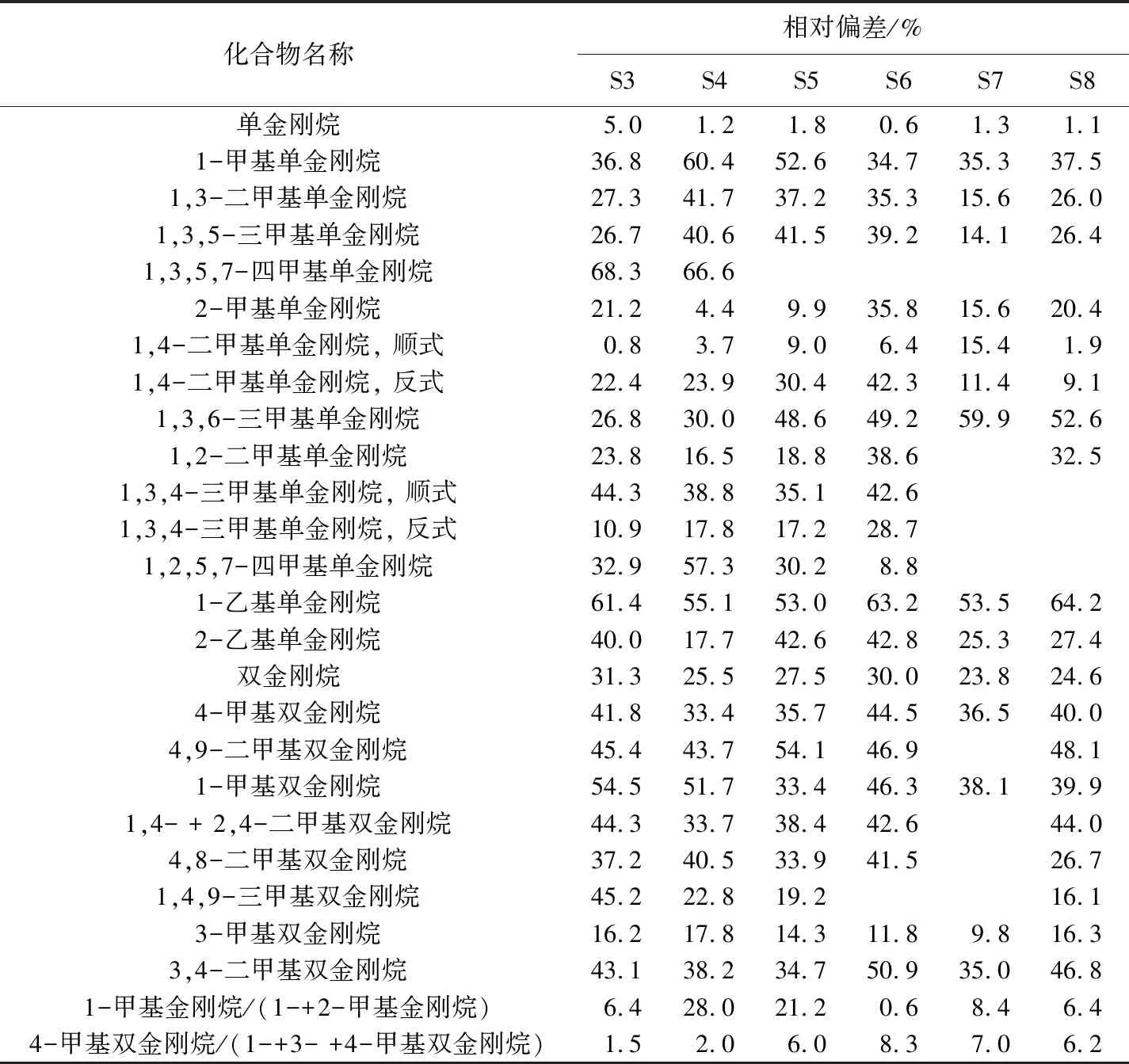
表4 GC-MS和GC×GC-FID定量結果的對比
注:表中相對偏差(Δ)的計算公式為Δ= |A1-A2|/(A1+A2), 其中A1表示GC×GC-FID的分析結果,
A2表示GC -MS的分析結果。
2.4 重復性
用細硅膠小柱分離S8樣品,用GC×GC-FID重復檢測7次,表5列出部分化合物的定量結果,7次重復試驗的相對標準偏差(RSD)小于5%,說明該方法重復性好,能滿足金剛烷系列化合物定量分析的要求。
3 結論
(1)由于標準物質的缺乏和共餾峰的影響,用GC-MS的定量金剛烷類化合物時,結果與真實值偏差較大。
(2)建立了用GC×GC-FID絕對定量常規金剛烷系列化合物的分析方法,該方法僅用一種氘代金剛烷就能定量其他結構的金剛烷系列化合物,且重復性好,值得推廣。
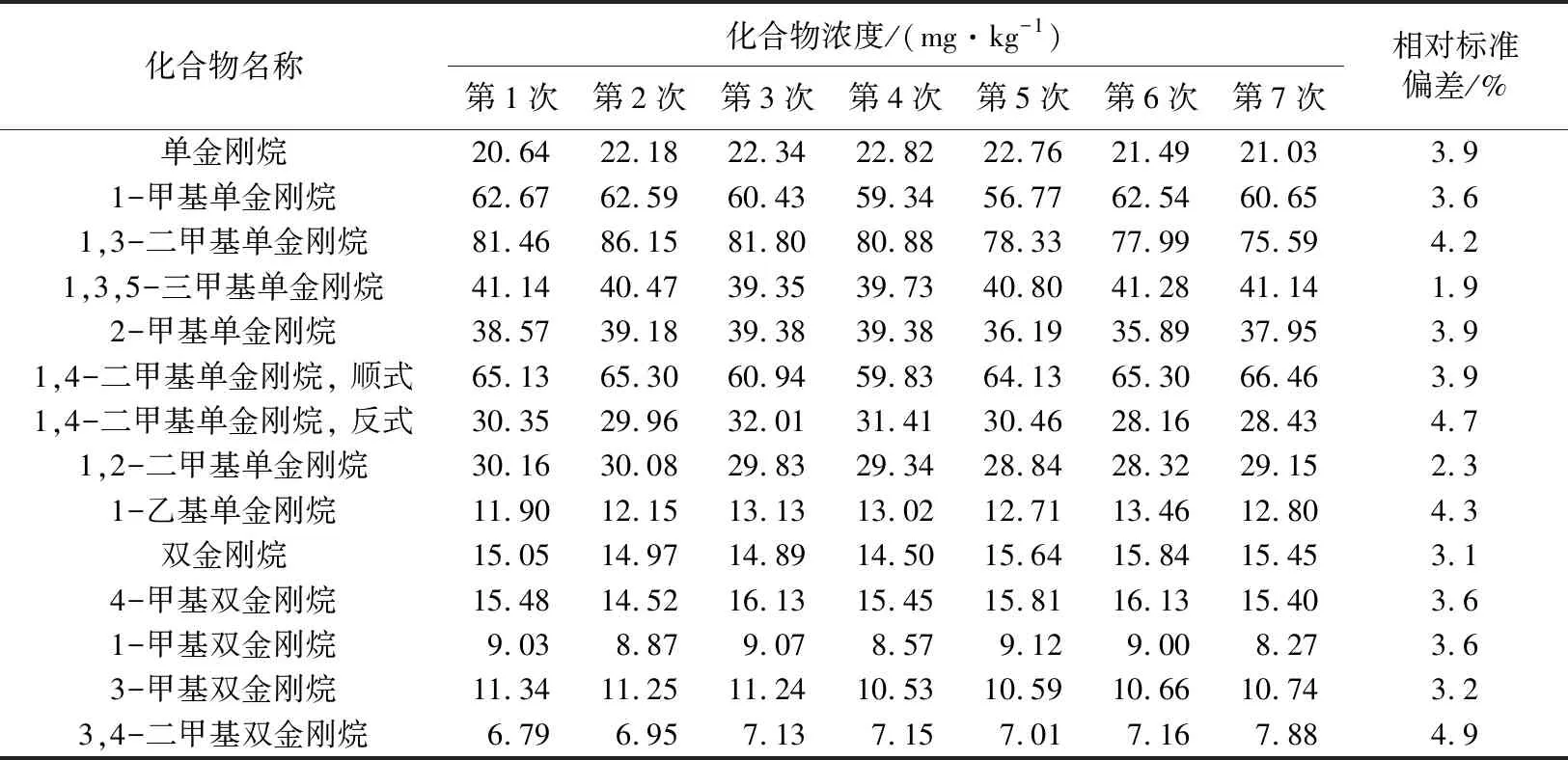
表5 S8樣品7次重復分析的定量結果
(3)建立了小柱分離的前處理方法,減少取樣量和溶劑消耗量,降低了金剛烷的揮發損失,適合取樣量少的樣品的分析。
(4)用GC×GC-FID定量分析金剛烷系列化合物的方法適合任何石油樣品,具有分離度高、無共餾化合物等特點,能夠得到客觀、準確的金剛烷系列化合物的定量結果,為該系列化合物的地球化學研究提供科學、有效的技術新手段。
猜你喜歡
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
兒童故事畫報(2019年5期)2019-05-26 14:26:14
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
