QuEChERS-氣相色譜-串聯質譜法快速檢測茶葉中6種禁用香精成分
2019-06-24 08:55:48王玉嬌鄧亞美王嘉琦花爾并天津科技大學生物工程學院天津300000中國檢驗檢疫科學研究院食品安全研究所北京100176
色譜 2019年7期
關鍵詞:檢測
王玉嬌, 劉 通, 鄧亞美, 王嘉琦, 陳 虹, 花爾并, 張 峰*(1.天津科技大學生物工程學院,天津 300000;.中國檢驗檢疫科學研究院食品安全研究所,北京 100176)
茶葉飲品被譽為世界三大飲料之一,我國是世界產茶大國,其產量和銷量位居前列。目前一些茶葉生產廠家受經濟利益驅動,將香精成分添加至茶坯中,改善和增強茶葉的香氣。研究表明,部分香精成分具有一定的毒性,比如香豆素具有致癌性,會導致肝退化與壞死、血管變化與海綿狀血管瘤,而側柏酮具有神經毒性,影響人類的身體健康[1-4]。還有一部分香精成分因為沒有確切的毒理報道而被認為對人體具有潛在威脅,因此我國食品安全管理部門、美國食品藥品管理局、歐洲食品安全局(EFSA)以及國際食用香料工業組織(IOFI)等對可用于食品的香精化學成分和含量都做出了明確限定[5]。我國食品安全國家標準GB 2760-2014《食品添加劑使用標準》中規定茶葉不得添加香精香料,但是目前茶葉中外加香精的檢測方法十分欠缺。香豆素、側柏酮、2-己基噻吩、反式-β-金合歡烯、p-薄荷-1,8(10)-二烯-9-乙酸乙酯和肉桂酸丁酯只能以天然生成的形式存在于某些食品,禁止作為添加劑用于茶葉。然而由于這6種禁用香精成分都是具有特定清香和芳香氣味的揮發性化合物,建立快速、準確的禁用香精成分檢測方法,對于保障茶葉質量安全和促進行業發展意義重大。
香精主要由揮發性化合物組成,用于食品中香精成分的定量和定性檢測方法主要有氣相色譜法(GC)[6]、氣相色譜-質譜法(GC-MS)[7-9]和氣相色譜-串聯質譜法(GC-MS/MS)[10-12]等。在復雜基質多組分分析時,氣相色譜法存在靈敏度低和檢測限高的缺點;GC-MS技術采用選擇離子監測(SIM)模式對化合物進行確認,基質復雜的樣品基質干擾大,易出現假陽性結果。GC-MS/MS是在GC-MS的基礎上增加了二級質譜信息,因此選擇性和抗干擾能力更強,定量更準確[13,14]。QuEChERS技術基于吸附原理,將吸附劑直接加入樣品提取液中,使之通過渦旋振蕩與基質干擾物充分接觸,將干擾物吸附在吸附劑上,達到樣品凈化的目的[15-17]。近年來,QuEChERS技術以其操作簡單、快速、高效、成本低廉等優點得到迅速發展,在食品領域得到了廣泛應用。
目前,針對這些香精的研究報道較少。譚麗容等[18]運用氣相色譜-質譜法檢測了薄荷香精中的限用物質反-2-己烯醛、α-己基肉桂醛、香豆素。周思等[19]運用固相萃取-氣相色譜-質譜法測定了植物飲料中測柏酮等9種植物毒素。其余化合物尚未見報道。本文采用QuEChERS前處理技術,同時結合GC-MS/MS技術,建立了針對茶葉中香豆素、側柏酮、2-己基噻吩、反式-β-金合歡烯、p-薄荷-1,8(10)-二烯-9-乙酸乙酯和肉桂酸丁酯的檢測方法。其中,QuEChERS技術操作簡單、快速、高效、成本低廉;GC-MS/MS靈敏度高,穩定性好,定性定量準確,抗基質干擾強。該法可為茶葉中禁用香精的精準測定提供技術支持。
1 實驗部分
1.1 儀器、試劑與材料
美國Agilent 7000C GC-MS/MS;GC配有7890A自動進樣器、多模式進樣口和DB-5MS毛細管色譜柱(30 m×0.25 mm×0.25 μm),數據采集由MassHunter完成;VORTEX KB-3渦旋振蕩器(江蘇海門其林貝爾儀器制造有限公司);3-30K高速冷凍離心機(德國Sigma公司)。
6種禁用香精標準品:側柏酮(thujone)、2-己基噻吩(2-hexylthiophene)、香豆素(coumarin)、肉桂酸丁酯(butyl cinnamate)、p-薄荷-1,8(10)-二烯-9-乙酸乙酯(p-menthol-1,8(10)-diene-9-ethyl acetate)(純度>96%,購自日本TCI公司),反式-β-金合歡烯(trans-beta-acacene,純度>98%,購自美國ChromaDex公司),乙酸乙酯、正己烷、二氯甲烷、丙酮(英國Fisher Scientific公司),乙二胺-N-丙基甲硅烷(PSA,德國Sigma-Aldrich公司)、石墨化碳黑粉末(GCB,德國CNW Technologies GmbH)、十八烷基鍵合硅膠(C18,美國Agilent公司)、無水硫酸鈉(成都市科龍化工試劑廠)。
1.2 標準溶液的配制
標準儲備液的配制:準確稱取6種香精標準品(10.0±0.1)mg,分別置于10 mL容量瓶中,加入少許乙酸乙酯,混勻,待標準品溶解再加入乙酸乙酯,定容至10 mL,可得1 g/L的儲備液,在-20 ℃冰箱中保存待用。
分別精密移取6種香精的標準儲備液100 μL至10 mL容量瓶中,用乙酸乙酯稀釋并定容至10 mL,可得10 mg/L混合標準溶液,于4 ℃冰箱中保存待用。
1.3 樣品前處理
準確稱取茶葉樣品1 g,置于50 mL離心管中,加入15 mL乙酸乙酯,渦旋1 min混勻,離心(25 ℃,8 000 r/min)5 min。將上清液15 mL加入第二個50 mL離心管中,然后加入10 mg無水硫酸鎂、5 mg C18和5 mg PSA,渦旋1 min,低溫離心5 min,用0.22 μm尼龍濾膜過濾后上機檢測。
1.4 基質標準曲線的制備
分別稱取不含目標物的空白茶葉樣品1 g(精確至0.01 g),按1.3節方法進行預處理。用基質提取液稀釋標準溶液,以峰面積(Y)為縱坐標對各被測組分的質量濃度(X,μg/L)為橫坐標作圖,繪制基質標準曲線。
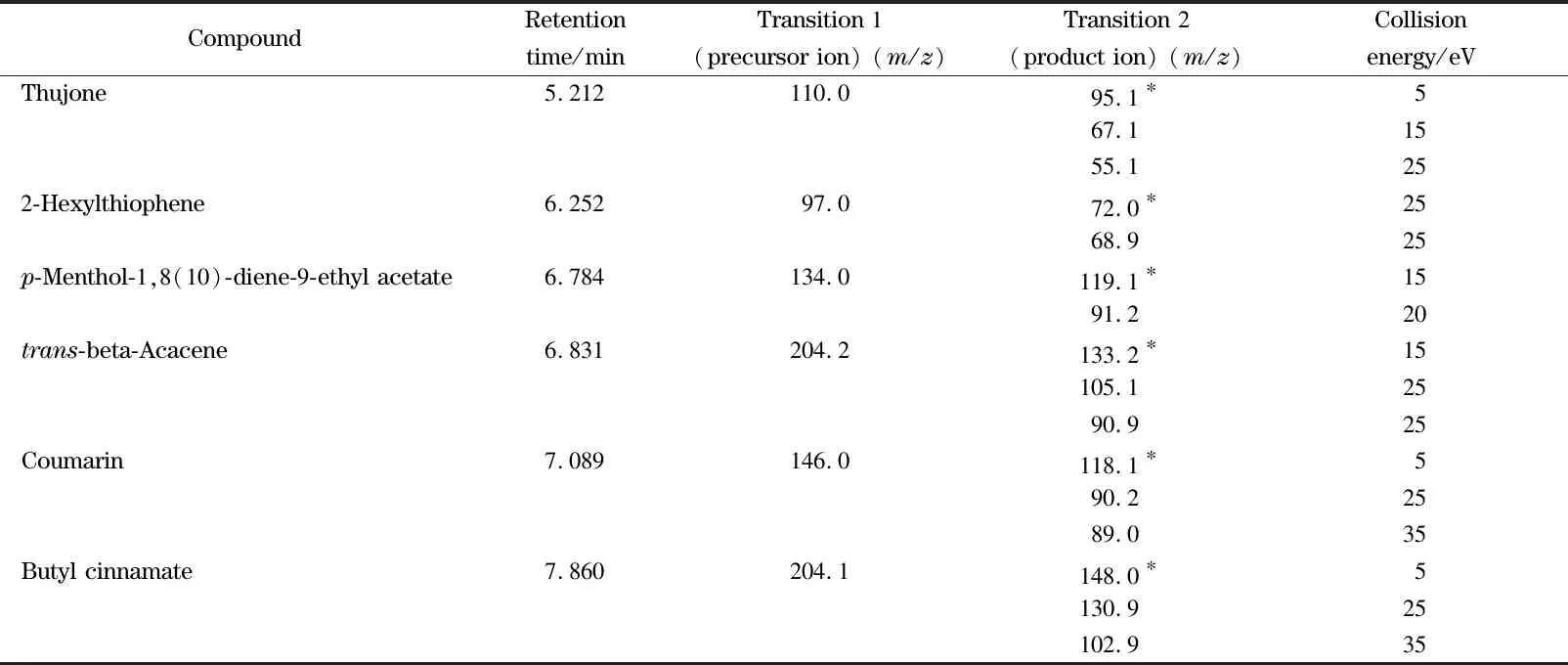
表1 6種化合物的保留時間和質譜參數Table 1 Retention time and MS parameters of the six compounds
* Quantitative ion.
1.5 GC-MS/MS條件
色譜條件:色譜柱DB-5MS(30 m×0.25 mm×0.25 μm),進樣口溫度250 ℃,不分流進樣;進樣量1.0 μL;載氣為氦氣(≥99.999%),流量1.0 mL/min。升溫程序:初始溫度40 ℃,保持1 min,以30 ℃/min升到220 ℃,保持2 min。
質譜條件:色譜-質譜接口溫度280 ℃;電子轟擊(EI)離子源;電子能量70 eV;多反應監測模式(MRM);溶劑延遲4 min。
2 結果與討論
2.1 色譜條件的考察
氣相色譜的升溫條件是非常重要的參數。從初始溫度40 ℃上升到220 ℃,本實驗考察了20、30、40 ℃/min 3種升溫速度時的情況。當升溫速度為40 ℃/min時,香豆素和反式-β-金合歡烯的保留時間相近,難以分開。20和30 ℃/min兩種升溫速度均可實現6種化合物的良好分離,為節約時間,最終選擇升溫程序為30 ℃/min。
2.2 質譜條件的考察
三重四極桿質譜條件的優化一般包括母離子、子離子以及碰撞能量等參數。在MRM參數優化過程中,先通過全掃描模式選取質荷比較大且相對豐度比較高的特征碎片作為母離子,然后將母離子進一步裂解,選取2個或3個碎片離子作為子離子,然后對每一對母離子/子離子以響應信號大小為依據優化碰撞能量。表1為所有目標化合物優化后的母離子、子離子和碰撞能量的信息。圖1為6種化合物的MRM色譜圖。
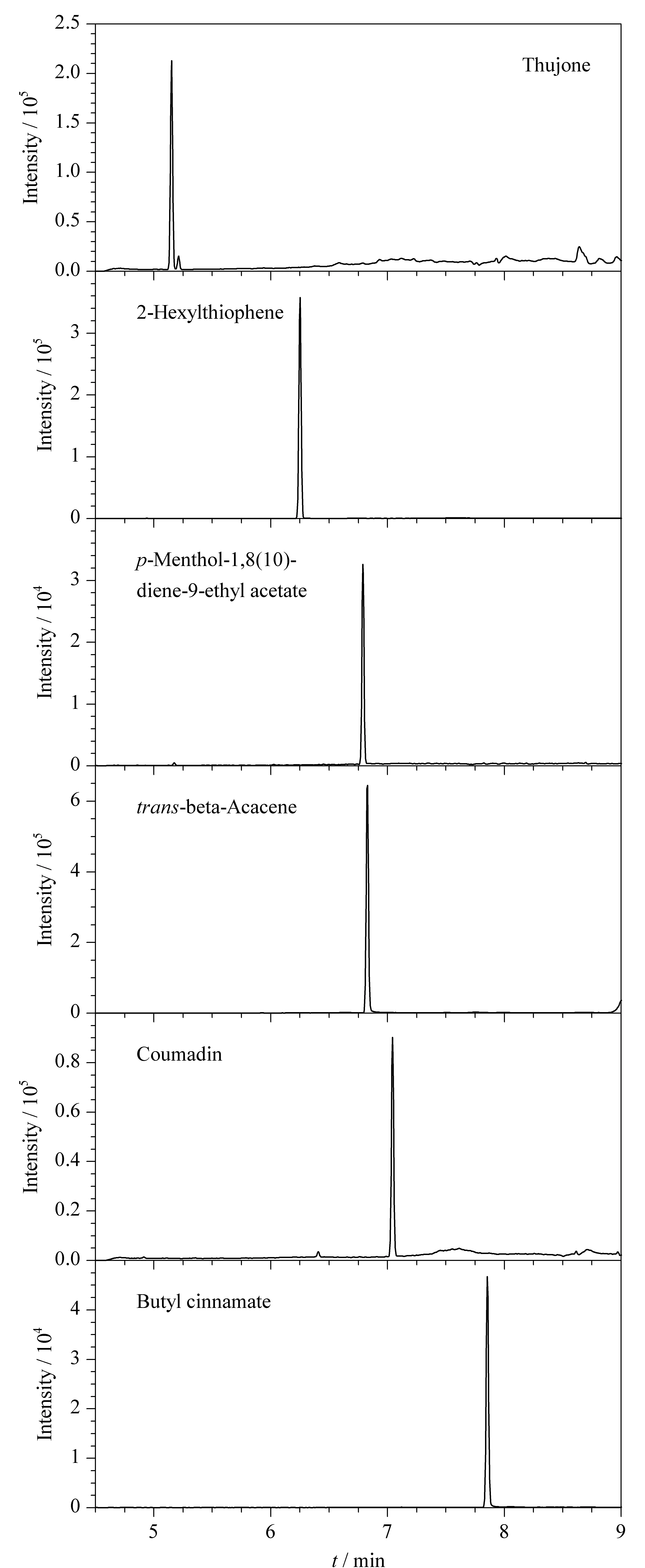
圖1 6種禁用香精化合物的MRM色譜圖Fig.1 MRM chromatograms of the six banned flavor compounds
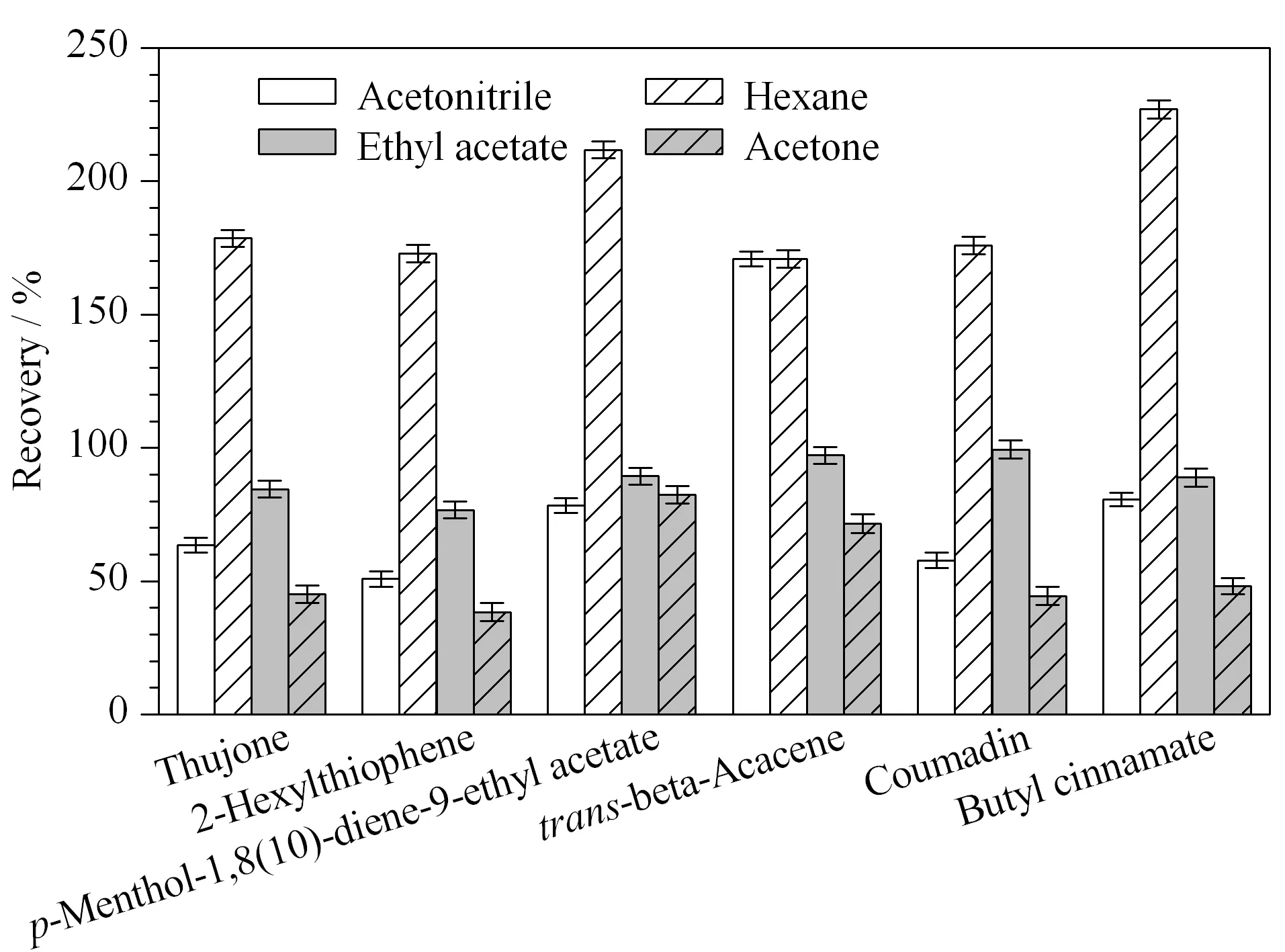
圖2 提取溶劑對6種禁用香精化合物回收率的影響(n=3)Fig.2 Effect of the extraction solvent on the recoveries of the six banned flavor compounds (n=3)
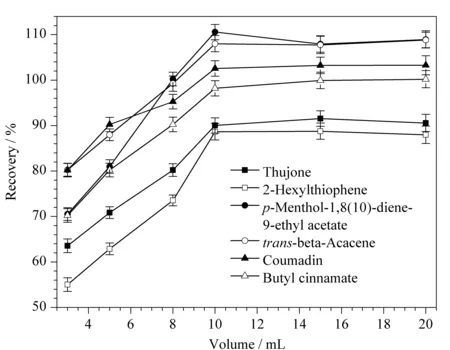
圖3 提取溶劑體積對6種禁用香精化合物回收率的影響(n=3)Fig.3 Effect of the volume of extraction solvent on the recoveries of the six banned flavor compounds (n=3)
2.3 QuEChERS前處理條件的優化
2.3.1提取溶劑的選擇
為了確定合適的提取溶劑,實驗以空白茶為基質,在30 μg/kg的加標水平下,分別以正己烷、乙腈、乙酸乙酯、丙酮為提取溶劑,比較6種目標物的提取效率,結果如圖2所示。當使用正己烷為提取溶劑時,6種化合物的回收率普遍偏高,回收率在170.85%~227.0%之間;當使用乙腈為提取溶劑時,2-己基噻吩的回收率只有50.89%,而反式-β-金合歡烯的回收率高達171.23%;使用丙酮為萃取劑時,除了p-薄荷-1,8(10)-二烯-9-乙酸乙酯回收率在正常范圍內(82.42%),其余5種化合物回收率偏低,在38.33%~71.58%之間;但當使用乙酸乙酯時,回收率全部在77.67%~99.42%之間,可滿足檢測要求,故最終選擇乙酸乙酯為最佳提取溶劑。
2.3.2提取溶劑體積的選擇
如圖3所示,以乙酸乙酯為提取溶劑,以加標水平30 μg/kg的空白茶葉為基質,以回收率為指標,考察不同提取溶劑體積(3、5、8、10、15和20 mL)對各目標物提取效率的影響。隨著提取溶劑體積的增大,目標物的回收率增加;當提取溶劑體積為10 mL時,回收率基本趨于穩定。為了確保回收率的穩定,最后選擇15 mL作為提取溶劑的用量。
2.3.3凈化方法的選擇
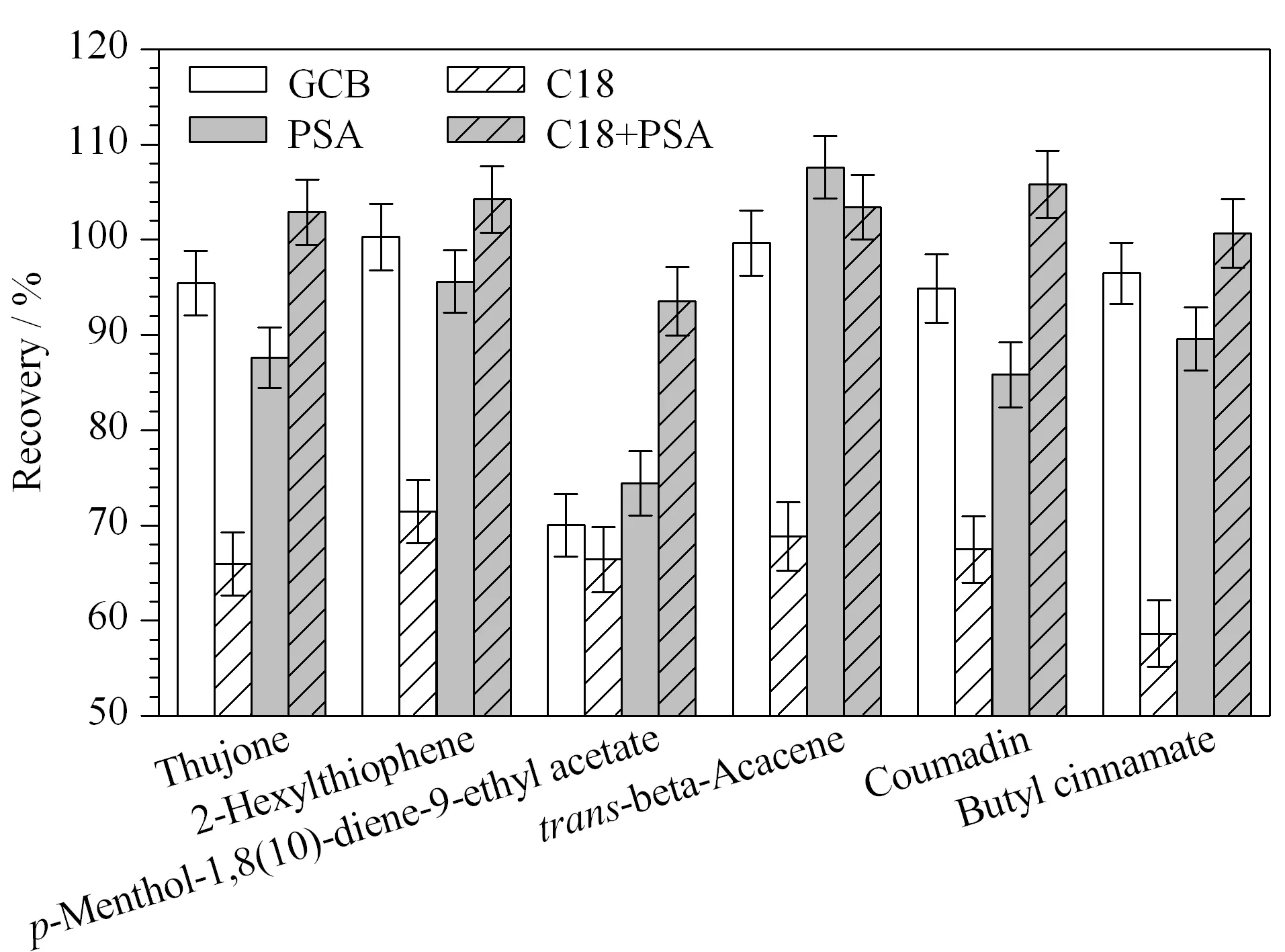
圖4 凈化材料對6種禁用香精化合物回收率的影響(n=3)Fig.4 Effect of the purification material on the recoveries of the six banned flavor compounds (n=3)
茶葉的基質復雜,含有色素、碳水化合物、茶多酚、脂類化合物等雜質。PSA、C18、GCB是QuEChERS前處理方法中常用的吸附劑。PSA可通過極性吸附作用、陰離子交換作用去除多酚、脂類等極性較強的雜質。C18對脂肪的去除效果明顯。GCB主要用于吸附色素。本實驗考察了分別加入10 mg GCB、10 mg C18和10 mg PSA,或5 mg PSA+5 mg C18吸附劑時的回收率。實驗結果如圖4所示:C18對目標化合物吸附性較強,回收率在72%以下;當使用GCB和PSA為凈化材料時,p-薄荷-1,8(10)-二烯-9-乙酸乙酯的回收率較低,分別為70.01%和74.40%;當同時使用PSA和C18時,凈化效果最好,化合物的回收率在93.52%~105.82%之間。另外,吸附劑用量是影響前處理凈化效果和回收率的重要因素。如果使用量小,凈化效果不明顯;如果使用量大,凈化效果明顯但回收率低,不能滿足檢測要求。通過實驗最終確定PSA用量為5 mg,C18用量為5 mg。
2.3.4無水硫酸鎂用量的優化
無水硫酸鎂作為脫水劑具有強大的吸水能力,與水結合后放出熱量,有利于目標物的提取。如無水硫酸鎂用量過少,會造成吸水不充分;其用量過多,會導致回收率偏低。因此有必要考察無水硫酸鎂的用量。在提取過程中,分別考察了5、10、20、30和40 mg無水硫酸鎂時的回收率,其他條件不變。結果如圖5所示,10 mg硫酸鎂能完全去除樣品中的水,提取回收率較合理;隨著硫酸鎂用量的增加,回收率逐漸降低。因此,最終選擇無水硫酸鎂的用量為10 mg。
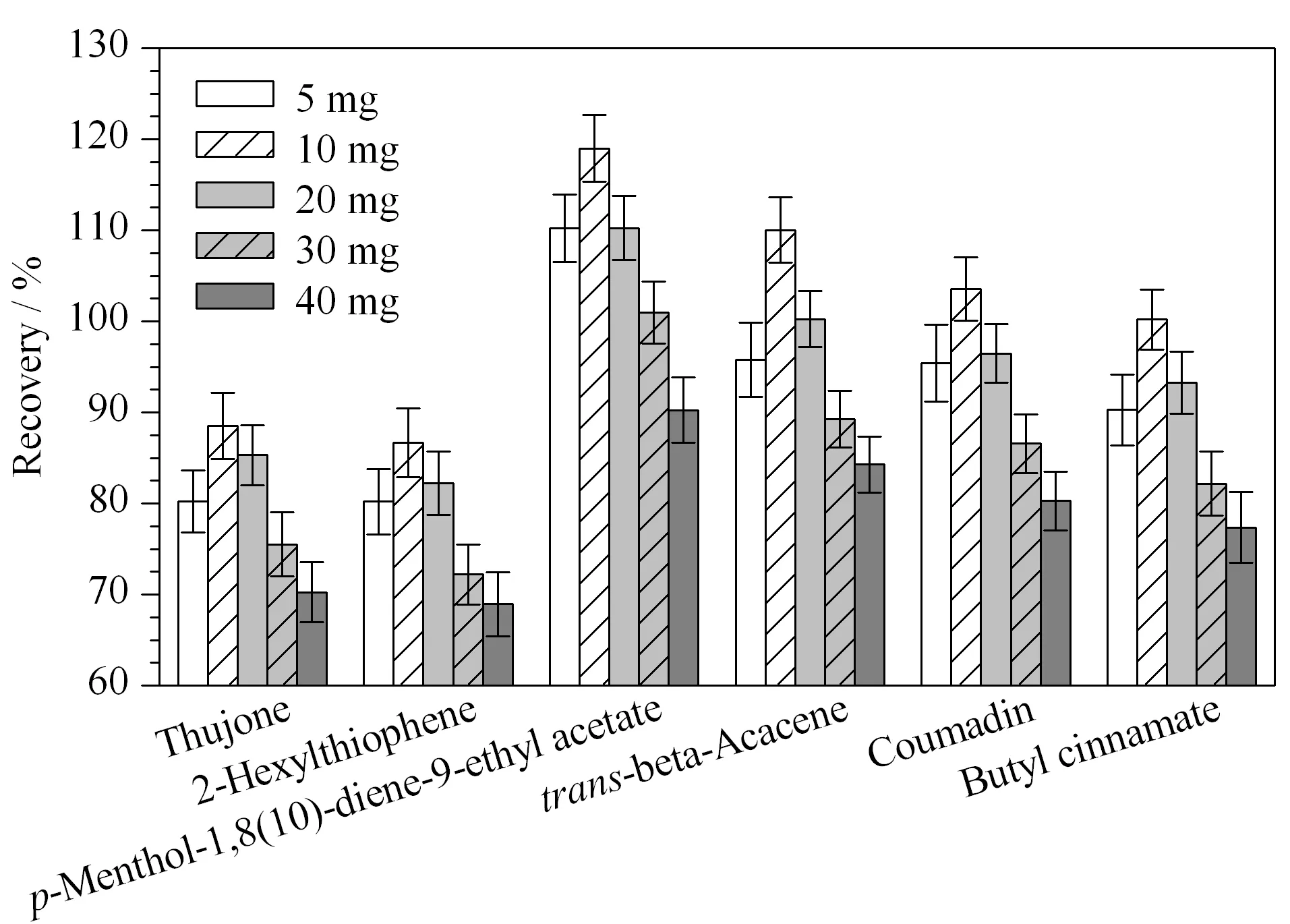
圖5 硫酸鎂用量對6種禁用香精化合物回收率的影響(n=3)Fig.5 Effect of the amount of magnesium sulfate on the recoveries of the six banned flavor compounds (n=3)
2.4 基質效應的考察
基質效應普遍存在于痕量物質的分析過程中,并會給定量結果造成影響。本實驗以基質匹配標準曲線與純溶劑標準曲線斜率之比來確定基質效應(ME)[20]:
ME=(1-Ss/Sm)×100%
(1)
其中,Ss是基質匹配曲線的斜率,Sm是標準曲線的斜率。當-50%

表2 6種禁用香精化合物的基質效應Table 2 Matrix effects (ME)of the six banned flavor compounds
2.5 方法驗證
2.5.1檢出限、定量限和線性范圍
配制6種化合物的基質標準溶液(1~200 μg/L),以峰面積(Y)為縱坐標,質量濃度(X,μg/L)為橫坐標,繪制標準曲線。6種化合物在1~200 μg/L范圍內呈良好的線性關系,其線性相關系數(R2)均大于0.999。在空白樣品中分別添加一系列濃度的混合標準溶液,進行前處理和測定,以3倍信噪比確定方法的檢出限(LOD),以10倍信噪比確定方法的定量限(LOQ)。如表3所示,各化合物的檢出限和定量限分別在0.005~0.1 μg/kg和0.02~2 μg/kg范圍內,表明該方法可以滿足復雜基質中痕量物質檢測的要求。其中側柏酮和香豆素的定量限分別是0.02 μg/kg和1 μg/kg,比文獻[18,19]報道中GC-MS的檢測下限(2.4 μg/kg和30 μg/kg)大幅降低,表明本方法有效地降低了茶葉中其他雜質和基質背景的干擾,具有更高的檢測靈敏度。
表36種禁用香精化合物的線性范圍、相關系數、檢出限和定量限
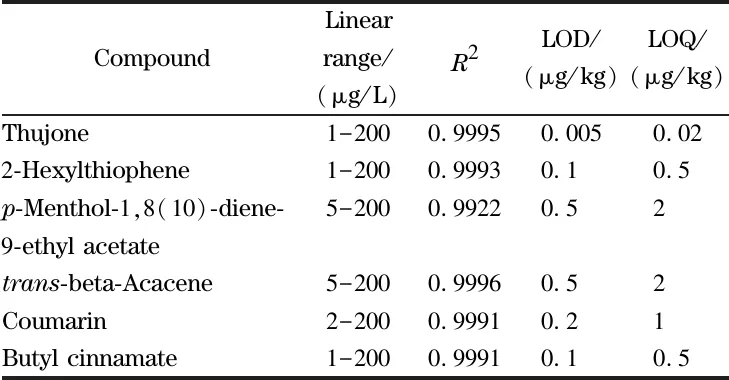
Table 3 Linear ranges,correlation coefficients (R2), limits of detection and limits of quantitation of the six banned flavor compounds
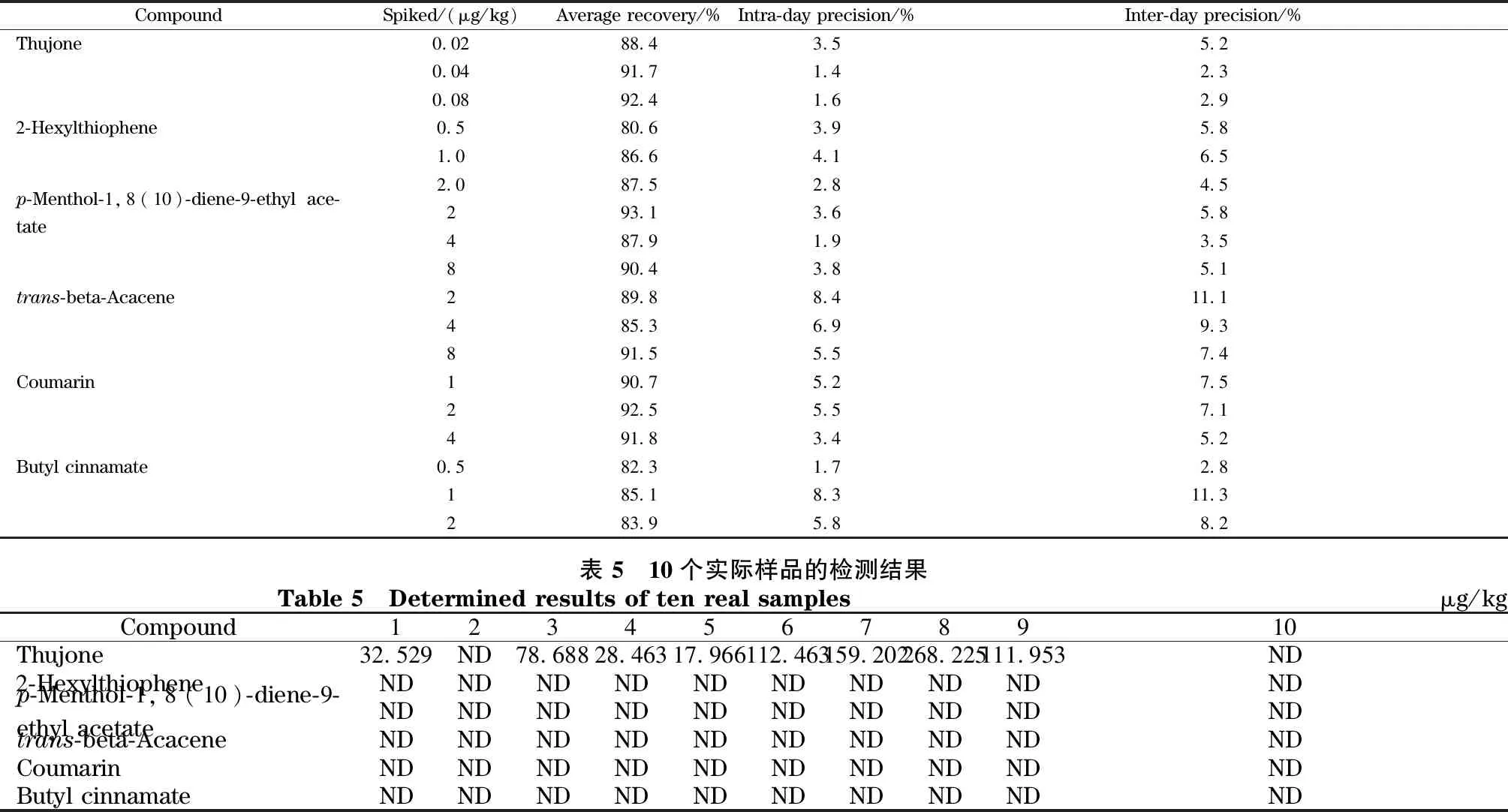
表4 6種禁用香精化合物在茶葉中的添加回收率、日內精密度和日間精密度(n=6)Table 4 Recoveries,intra-day precisions and inter-day precisions of the six banned flavor compounds spiked in tea (n=6)
ND:not detected.
2.5.2回收率和精密度
取空白茶葉樣品1 g,分別添加3個水平(1、2、4倍定量限)的標準儲備液,進行加標回收試驗,每個水平做6次平行,計算平均回收率和日內、日間精密度。結果如表4所示,6種化合物的平均回收率是82.3%~93.1%,日內精密度在1.4%~4.1%之間,日間精密度在2.3%~6.5%之間。
2.5.3實際樣品檢測
針對10個市售的茶葉樣品,用本文建立的方法進行檢測。結果如表5所示,2-己基噻吩、p-薄荷-1,8(10)-二烯-9-乙酸乙酯、反式-β-金合歡烯、香豆素、肉桂酸丁酯5種化合物在實際樣品中均未檢出,而側柏酮則在其中的8個樣品中檢出,含量最高達到268.225 μg/kg(色譜圖見圖6)。我國目前尚未有茶葉中側柏酮含量的限量標準。歐盟法規EC 1334/2008條例中規定,青蒿生產的飲料中側柏酮含量不得高于0.5 mg/kg(不含酒精飲料)和35 mg/kg(含酒精飲料),非青蒿生產的飲料中側柏酮含量不得高于10 mg/kg(含酒精飲料)。側柏酮有可能是茶葉樣品中的內源性天然成分,有必要對其來源開展進一步研究。
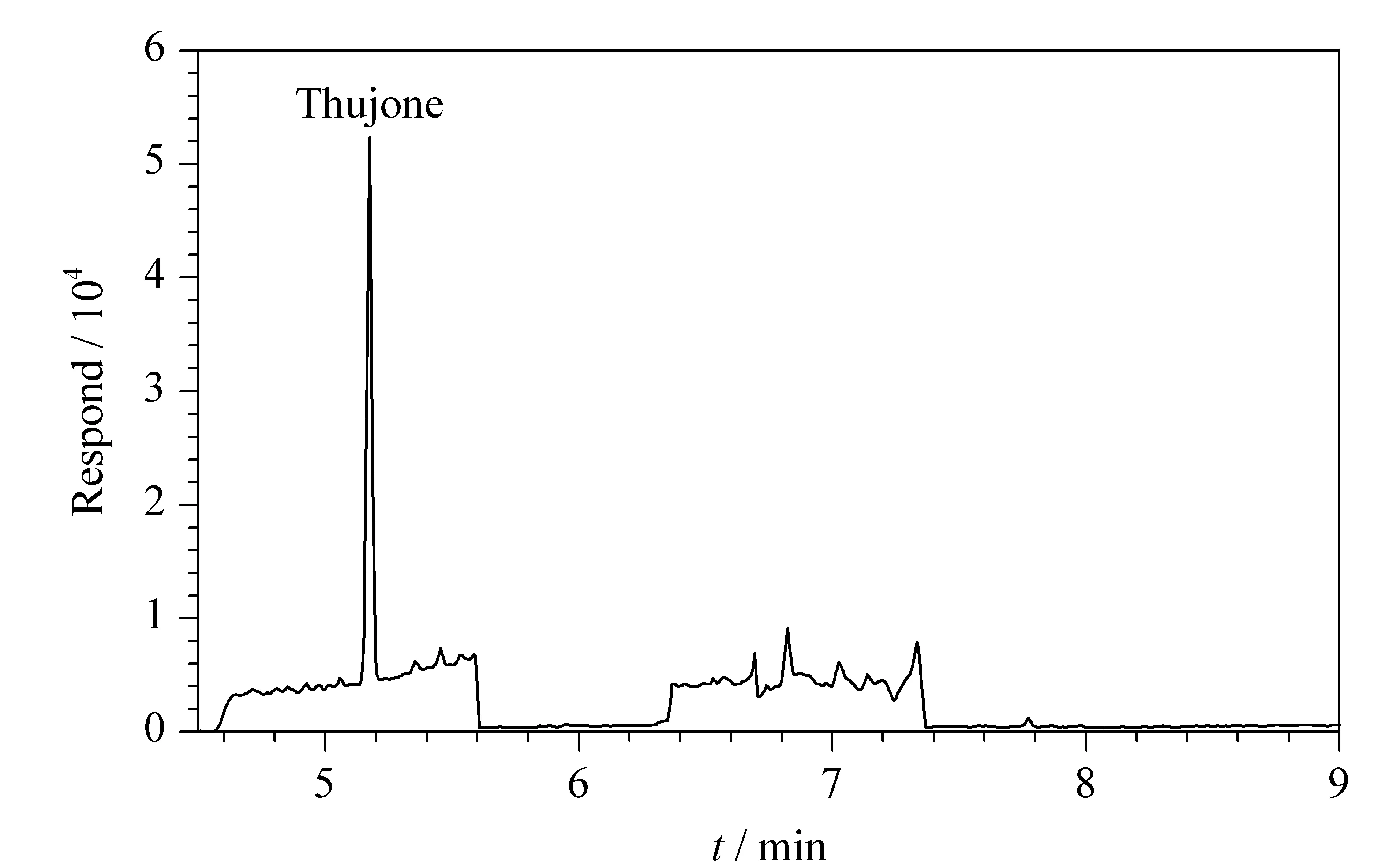
圖6 陽性樣品的色譜圖Fig.6 Chromatogram of a positive sample
3 結論
本文利用QuEChERS技術同步提取和凈化茶葉樣品,結合GC-MS/MS建立了快速測定茶葉中6種禁用香精成分的分析方法。方法學評價和實際樣品檢測結果表明,該方法操作簡便,靈敏度高,凈化效果較好,可用于茶葉中多種禁用香精的檢測。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
