Neonatal cholestasis and hepatosplenomegaly caused by congenital dyserythropoietic anemia type 1: A case report
2019-06-20 07:59:52CatalinaJaramilloAnnaErmarthAngelicaPutnamMarkDeneau
World Journal of Hepatology 2019年5期
關鍵詞:規范
Catalina Jaramillo, Anna K Ermarth, Angelica R Putnam, Mark Deneau
Catalina Jaramillo, Anna K Ermarth, Mark Deneau, Department of Pediatrics, University of Utah,Salt Lake City, UT 84113, United States
Angelica R Putnam, Department of Pathology, University of Utah, Salt Lake City, UT 84113,United States
Abstract
Key words:Congenital dyserythropoietic anemia; Hemochromatosis; Pulmonary hypertension; Jaundice; Case report
INTRODUCTION
Congenital dyserythropoietic anemia type 1 (CDA1) is an autosomal recessive disorder of ineffective erythropoiesis, resulting in increased iron storage, and considered a form of secondary hemochromatosis[1]. Most CDA1 patients have a mutation in the CDAN1 gene[2]. CDA1 is usually diagnosed in children and adolescents with moderate to severe macrocytic anemia. However, it can rarely present in the neonatal period with severe anemia at birth[2]. Additional clinical findings include hepatosplenomegaly (HSM), jaundice, cholestasis, liver dysfunction,transient thrombocytopenia and persistent pulmonary hypertension of the newborn[2].The diagnosis is based on hematologic abnormalities and positive genetic testing[3].Bone marrow biopsy findings include spongy heterochromatin, enlargement of nuclear pores and invagination of cytoplasm into the nuclear area[4].
Prior reports have described liver biopsy findings of extramedullary hematopoiesis and iron accumulation in autopsies and adult patients[5-8]. There have been no prior reports of neonatal liver histologic findings of CDA1. We report a case of CDA1 in a newborn presenting with severe anemia, cholestasis and liver failure, where liver biopsy helped confirm the diagnosis.
CASE PRESENTATION
Chief complaints
This is a former 37 wk and 3 d old female transferred to our institution due to respiratory failure.
Birth history
The patient was delivered by emergency Cesarean section due to non-reassuring fetal heart rate tracings at an outside hospital to a 28-year-old, Caucasian, gravida 4, para 2,0, 1, 2 with an unremarkable pregnancy. A prior pregnancy was significant for fetal demise at 35 wk without autopsy or known etiology for the fetal demise. Parents are both healthy and there is no history of consanguinity. Perinatal laboratory results included maternal blood type O (+) with negative antibody screen, negative venereal disease research laboratory, hepatitis B, and human immunodeficiency virus and rubella. Apgar scores were 7 and 8. Birth weight was 3070 g (21stpercentile), length 18 inches (16thpercentile), occipital frontal circumference 32.5 cm (3rdpercentile).
Physical examination upon admission
At birth, this patient had no facial or limb dimorphism. She was started on supplemental oxygen due to duskiness 10 min after birth. Subsequently, she required endotracheal intubation and initiation of inhaled nitric oxide. She was then transferred to our institution due to respiratory failure on day of life (DOL) 1. On arrival, she was found to have HSM.
Laboratory examinations
On admission to our institution, she was found to have liver dysfunction with an International normalized ratio (commonly referred to as INR) of 2.1, total bilirubin of 9 mg/dL, direct bilirubin of 2.1 mg/dL, aspartate aminotransferase 655 U/L andalanine aminotransferase 65 U/L. Partial thromboplastin time was within normal limits, with mildly low fibrinogen and elevated D-dimers. Anemia and thrombocytopenia were also present. The anemia was present since birth with a hemoglobin of 7.4 g/dL and hematocrit of 23.7%. Her platelets were initially normal but soon started to decline, with a nadir of 54 k/μL on DOL 1. She was also found to have pulmonary hypertension, right ventricular hypertrophy and required high frequency oscillator ventilation due to hypoxemic respiratory failure.
Additional laboratory work-up included serum ferritin of 40664 ng/mL and normal soluble interleukin 2 receptor. Initially her gamma-glutamyltransferase(commonly referred to as GGT) was normal and then peaked at 390 U/L on DOL 25.Infectious studies included negative herpes simplex virus, Epstein-Barr virus,cytomegalovirus, adenovirus, parvovirus, enterovirus, echovirus, parechovirus and human herpesvirus 6 PCR. Multiple blood cultures and urine cultures were also negative. She also had negative work-up for inborn errors of metabolism:normal serum plasma amino acids, urine organic acids, ammonia and very long/branchedchain fatty acids.
Imaging examinations
Imaging studies included an initial echocardiogram on DOL 1, which showed a large patent ductus arteriosus, small patent foramen ovale and dilated and hypertrophied right ventricle with suprasystemic pressures. An abdominal ultrasound showed HSM with minimal ascites, and a liver Doppler was normal.
Further diagnostic work-up
A salivary gland biopsy performed on DOL 5 did not show any evidence of iron deposition. On DOL 15, a liver biopsy was performed, which showed iron deposition(Figure 1) and erythroblasts with spongy appearance (Figure 2). Immunohistochemical staining for cytomegalovirus and HSV were negative. A genome rapid sequencing panel of over 4500 genes was performed (ARUP Laboratories, Salt Lake City, UT, United States) and revealed novel compound heterozygous variants inCDAN1, c.2174G>A (p.Arg725Gln) and c.1003C>T (p.Arg335Trp), each variant inherited from an asymptomatic parent.
FINAL DIAGNOSIS
The final diagnosis of the presented case is CDAN1 resulting from c.2174G>A(p.Arg725Gln) and c.1003C>T (p.Arg335Trp) mutations.
TREATMENT
The infant remained on broad spectrum antibiotics, antivirals and required multiple packed red blood cell, fresh frozen plasma and platelet transfusions. She also received intravenous immunoglobulin. She was weaned off mechanical ventilation on DOL 16 and was discharged from the neonatal intensive care unit (known as the NICU) at DOL 43.
OUTCOME AND FOLLOW-UP
Her INR normalized by DOL 2. Her ferritin levels remained elevated but were declining with a level of 4133 ng/mL at NICU discharge. She had improving liver enzymes, bilirubin and thrombocytopenia throughout her NICU stay. At discharge,she was on nasal cannula and sildenafil for persistent pulmonary hypertension. By 7 wk of age, her bilirubin had normalized, and by 4 mo of age, her liver enzymes and GGT had normalized. At her first gastroenterology follow-up 4 wk after discharge,her organomegaly had resolved. At 1 year of age, her ferritin level had decreased to 1139 ng/mL and had 9.3 mg of iron/g of liver tissue determined by magnetic resonance hepatic iron quantification, still consistent with iron overload[5]. She remains transfusion-dependent.
由表1可知,大壩上游壩坡在各種運行工況下穩定性較差,均小于規范允許值;特別是在工況5,上游壩坡安全系數僅有0.98。說明上游壩坡存在失穩可能,而下游壩坡穩定性較好,因此需重點加固上游壩坡。
DISCUSSION
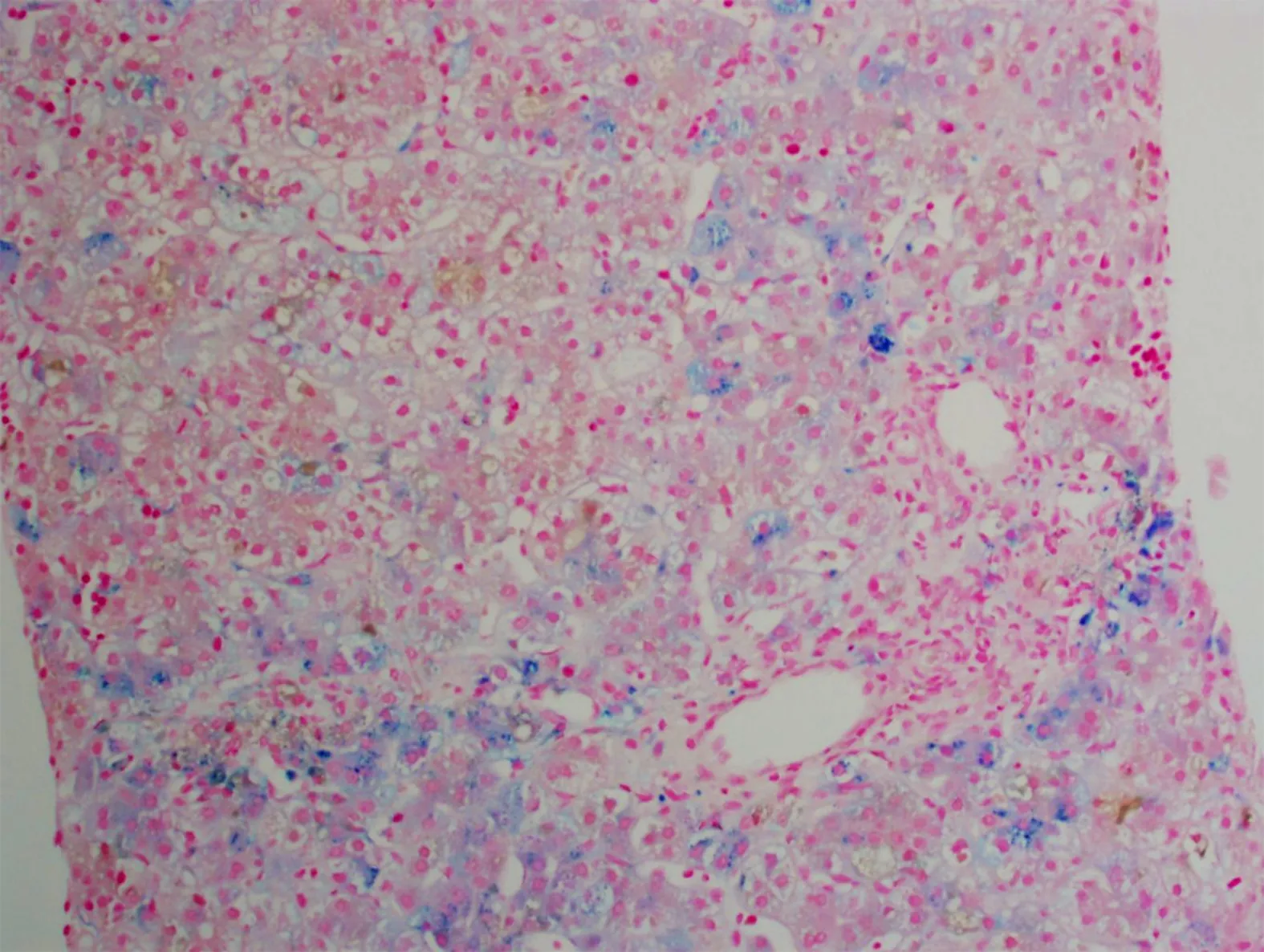
Figure 1 Approximately 10%-15% of the hepatocytes contained iron granules.
CDA1 is a rare disorder of ineffective erythropoiesis that leads to severe anemia[6]. It has been mainly described in European countries and in the Bedoiun Israeli population[2]. The diagnosis is more commonly suspected in patients presenting with hematologic abnormalities such as moderate-severe macrocytic anemia (MCV > 90),inappropriately low reticulocytes for degree of anemia, macrocytosis, elliptocytes and basophilic stippling on peripheral blood smear, bone marrow aspirate findings of erythroid hyperplasia with interchromatic bridges on light microscopy and erythroblasts with spongy appearance of heterochromatin and invaginations of the nuclear membrane on electron microscopy[3]. Other common findings may include jaundice, splenomegaly, limb dimorphism, hypoplastic nails and syndactyly[3].
Diagnosis in the newborn period is rare[2]. The patient described above presented with multiple clinical characteristics previously described in the literature. HSM and early jaundice are commonly encountered (65% and 53%, respectively). Neonates can also present with direct hyperbilirubinemia in up to 20% of cases. Thrombocytopenia has been described as transient, which is consistent with this patient's presentation[2].Persistent fetal circulation/pulmonary hypertension has also been reported in up to 15% of patients; the reported cases have had pulmonary hypertension without any underlying cardiopulmonary abnormalities requiring high pressure ventilation[2,7]. It is also reported that patients who have clinical manifestations of CDA1 in the neonatal period have severe intrauterine anemia at birth, and there have been cases of hydrops fetalis[8,9].
In this case, a liver biopsy supported evidence for the diagnosis of CDA1 before genetic testing was performed, with identification of typical siderosis and extramedullary hematopoiesis[6,10]. Both of these findings are consistent with prior adult liver pathology reports. Most recently in 2016, Salihogluet al[10]reported a case of CDA1 in an adult in whom a liver biopsy was performed to exclude Wilson’s disease and was found to have extramedullary hematopoiesis. Another case report describes a 28-year-old diagnosed with CDA1 with a liver biopsy that showed massive siderosis and early cirrhosis[6]. Autopsy reports have also been described with findings of extramedullary hematopoiesis of the liver and spleen[8].
The initial therapeutic modality for this disease is intermittent blood transfusions,however if patients become transfusion-dependent, there have been cases of successful treatment with interferon alpha[6,11,12]. Up to 80% of affected neonates require blood transfusions in the first month of life, with reported transfusion independence by 4 mo of age in 88% of patients[2]. There are also three reported cases of bone marrow transplantation in patients resistant to interferon therapy[3,13]. Our patient is currently receiving intermittent blood transfusions approximately every 4 wk.
From a gastrointestinal and hepatology standpoint, CDA1 patients have a future risk of gallstones (reported in patients as young as 4 years of age)[4]and secondary hemochromatosis; the latter develops with age due to increased iron absorption even in patients who are not chronically transfused[3]. Hence, it is suggested that patients are periodically monitored (every 3 mo) for iron overload starting at age 10[3].
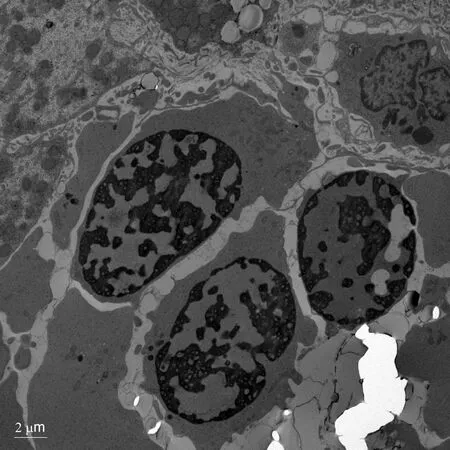
Figure 2 Electron microscopy showing erythroblasts with dense heterochromatin and translucent vacuoles.
CONCLUSION
Liver biopsy can be a helpful tool in the diagnosis of infants with unexplained liver dysfunction. This case report describes the liver histopathology and electron microscopy findings of CDA1 caused by a novel genetic mutation in the pediatric age group. CDA1 is in the differential diagnosis of infants with unexplained anemia,hyperbilirubinemia and HSM.
猜你喜歡
中小學教師培訓(2022年11期)2022-11-01 03:13:54
中小學教師培訓(2022年10期)2022-10-15 02:18:04
保健醫苑(2022年6期)2022-07-08 01:24:52
北部灣大學學報(2022年1期)2022-06-22 04:58:38
北部灣大學學報(2022年2期)2022-06-21 11:44:36
中國信息化(2022年4期)2022-05-06 21:24:05
北部灣大學學報(2021年1期)2022-01-27 06:40:10
現代儀器與醫療(2021年4期)2021-11-05 08:25:08
北部灣大學學報(2021年6期)2021-06-21 06:01:48
北部灣大學學報(2021年4期)2021-04-28 08:01:04
 World Journal of Hepatology2019年5期
World Journal of Hepatology2019年5期
- World Journal of Hepatology的其它文章
- Successful treatment of noncirrhotic portal hypertension with eculizumab in paroxysmal nocturnal hemoglobinuria: A case report
- Carvedilol vsendoscopic variceal ligation for primary and secondary prevention of variceal bleeding: Systematic review and metaanalysis
- Expanding etiology of progressive familial intrahepatic cholestasis
- Hepatitis C virus antigens enzyme immunoassay for one-step diagnosis of hepatitis C virus coinfection in human immunodeficiency virus infected individuals
- Hepatitis C virus cure with direct acting antivirals: Clinical,economic, societal and patient value for China
- Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis