中空碳納米棒負載Fe3O4的制備及催化性能研究
2019-05-28 02:41:34孟慶男崔娟妮
西安理工大學學報 2019年1期
孟慶男,崔娟妮,趙 康
(西安理工大學,材料科學與工程學院,陜西西安710048)
近年來,隨著經濟的不斷發展,我國的水污染問題日益嚴重,對國民健康和社會發展構成了嚴重威脅。Fenton技術是利用H2O2分解產生的 ·OH來降解水中有機污染物的一種方法,由于其綠色、高效,受到了研究人員的廣泛重視[1]。Fe3O4納米材料作為一種常用的非均相Fenton反應催化劑,不僅活性高,還克服了均相Fenton反應中易產生鐵污泥、鐵鹽無法重復利用等缺點。但磁性納米粒子存在不穩定、易團聚、容易被氧化等缺點,限制了其催化性能的發揮[2]。目前,常用的方法是將Fe3O4負載在惰性載體中。對于載體,不但要起到防止Fe3O4納米材料聚集的作用,還需保證水中有機污染物能到達催化劑表面[3-6]。
碳在常溫下非常穩定,能夠耐強酸、強堿,與有機物及氧化物相比更適合作為納米催化劑的載體[3]。此外,中空結構碳材料具有比表面積大、密度小,以及良好的吸附性和通過性,已被廣泛應用于催化領域[7]。因此,本文選取商用Fe2O3納米棒為前驅體,利用“一鍋法”實現二氧化硅和間苯二酚-甲醛樹脂的包覆,隨后通過熱處理同時實現樹脂殼層的碳化和內部Fe2O3的還原,最終制備中空碳納米棒負載Fe3O4材料(Fe3O4@h-C)。所制備的Fe3O4@h-C在亞甲基藍的催化降解反應中顯示出良好的催化活性和重復使用性能。中空結構不但有利于阻止納米Fe3O4的聚集,還能保證其與有機污染物充分接觸,有利于Fe3O4材料催化性能的提升。
1 實驗
1.1 試劑與儀器
Fe2O3納米棒,購自阿拉丁試劑公司。無水乙醇、氨水(質量分數25%)、正硅酸乙酯(TEOS)、間苯二酚、乙醇、甲醛、NaOH均為分析純,購自國藥集團化學試劑有限公司。實驗用水為去離子水。
JEOL-2010型透射電子顯微鏡(TEM),日本電子株式會社;Nicolet Avatar 360型紅外光譜儀(FTIR),PerkinElmer公司;Lambda 750型紫外-可見分光光度計(UV-Vis),PerkinElmer 公司;SiemensD-5005型X射線衍射儀(XRD),Bruker公司;DTG-60H型熱重分析儀(TGA),日本島津公司。
1.2 催化材料的制備
稱取0.479 g Fe2O3納米棒,加入到32 mL去離子水、128 mL乙醇和3 mL氨水的混合液中,攪拌30 min后加入1 mL正硅酸乙酯(TEOS)。10 min后,向溶液中加入0.2 g間苯二酚,再隔10 min加入0.28 mL甲醛。磁力攪拌8 h后,將產物離心、洗滌、干燥,所得產物在5 %氫氣、95 %氮氣的氣氛中500 ℃還原、碳化,保溫2 h。最后用1 mol/L的NaOH刻蝕SiO2中間層,得到Fe3O4@h-C納米棒。作為對比,在制備過程中不加TEOS制備四氧化三鐵-碳核殼型(Fe3O4@C)納米棒;在制備過程中不加間苯二酚和甲醛制備純Fe3O4納米棒。
文章中關于粒徑和尺寸的數據,均通過Photoshop軟件測量TEM照片得到,每次測量不少于20組數據,然后取平均值。
1.3 催化性能測定
將200 mL(50 mg/L)亞甲基藍溶液調節到指定溫度和pH 值,然后稱取20 mg的催化劑樣品分散于溶液中,攪拌30 min至吸附平衡,加入一定量的雙氧水溶液(30%);通過紫外-可見分光光度計對亞甲基藍溶液在665 nm處的吸光度進行檢測,并計算其降解率[1]。
2 結果與討論
2.1 催化材料的結構與形貌
圖1是在Fe2O3納米棒表面包覆二氧化硅和間苯二酚-甲醛樹脂后,產物在氮氣下的TGA測試結果。從曲線上可以看出,失重是分兩個階段的。第一階段是200 ℃以下,這段的失重主要是因為樣品中的少量水分和其他揮發性的小分子雜質的損失;第二階段是從200 ℃開始的,在這個階段,樹脂逐漸碳化,當溫度上升到800 ℃以上時,曲線趨于直線,此時樣品的失重率約為17%[8]。根據TGA曲線,樣品的熱處理溫度選擇500 ℃,既可保證樹脂的碳化,又能防止Fe3O4在高溫下轉化為單質鐵[9]。
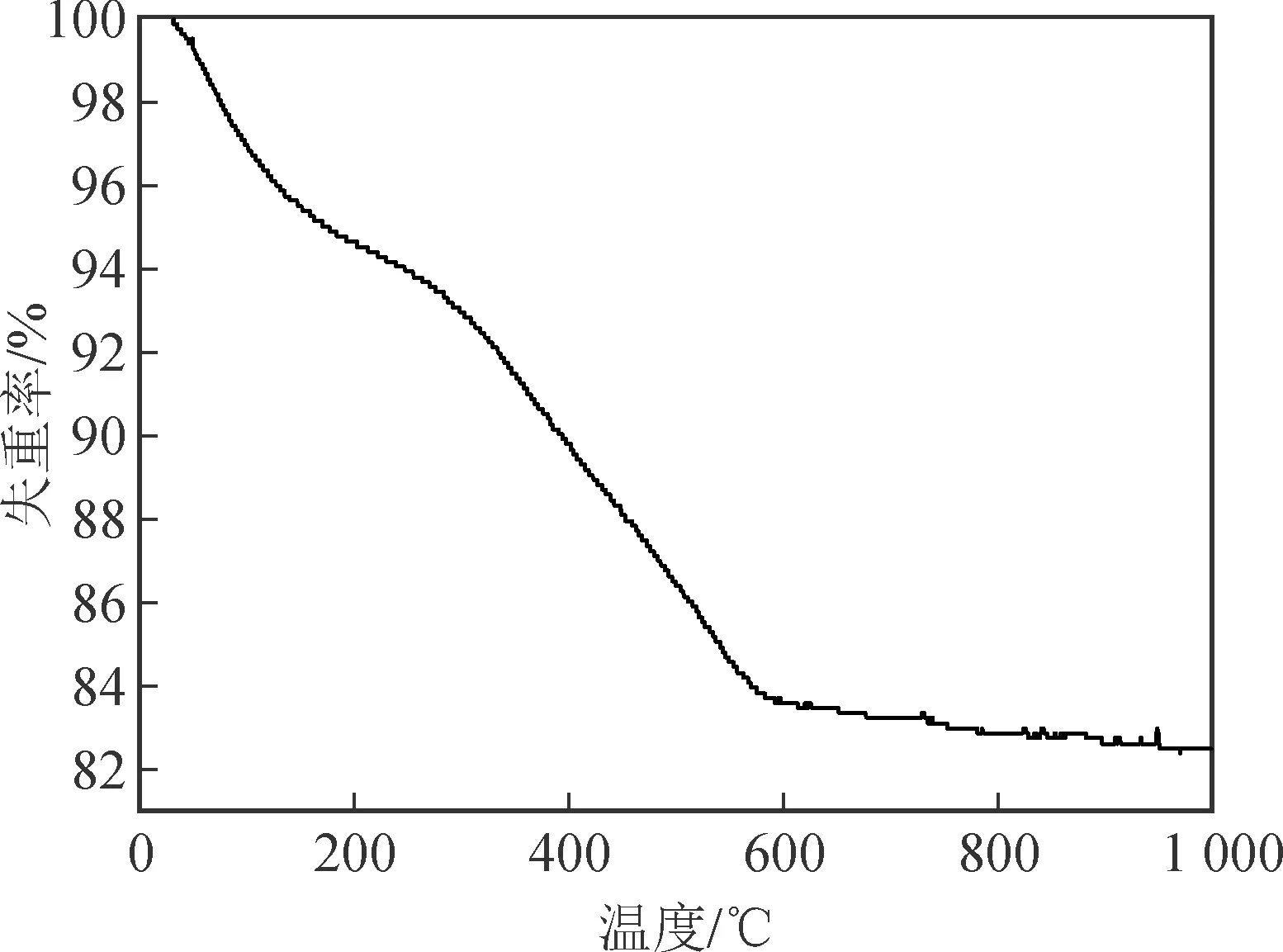
圖1 包覆產物的TGA曲線Fig.1 TGA curve of the coated Fe2O3 precursor
圖2(a)為所采用的商用Fe2O3納米棒的XRD圖譜,其衍射峰與斜方六面體晶型α-Fe2O3的標準卡片(JCPDS No.87-1164)相符,沒有雜質相,說明其純度較高[10]。經過熱處理及NaOH刻蝕后,最終產物的XRD譜圖(圖2(b))中,在2θ為 30.1°、35.7°、43.3°、57.2°、62.8°處的衍射峰分別對應面心立方Fe3O4晶體(JCPDS No.75-0033)的(220)、(311)、(400)、(511)、(440)晶面,這表明Fe2O3已完全轉化為Fe3O4,且在刻蝕的過程中沒有發生明顯的氧化[11]。此外,譜圖中沒有出現石墨的衍射峰,說明碳化溫度較低,碳以無定型形式存在[9]。
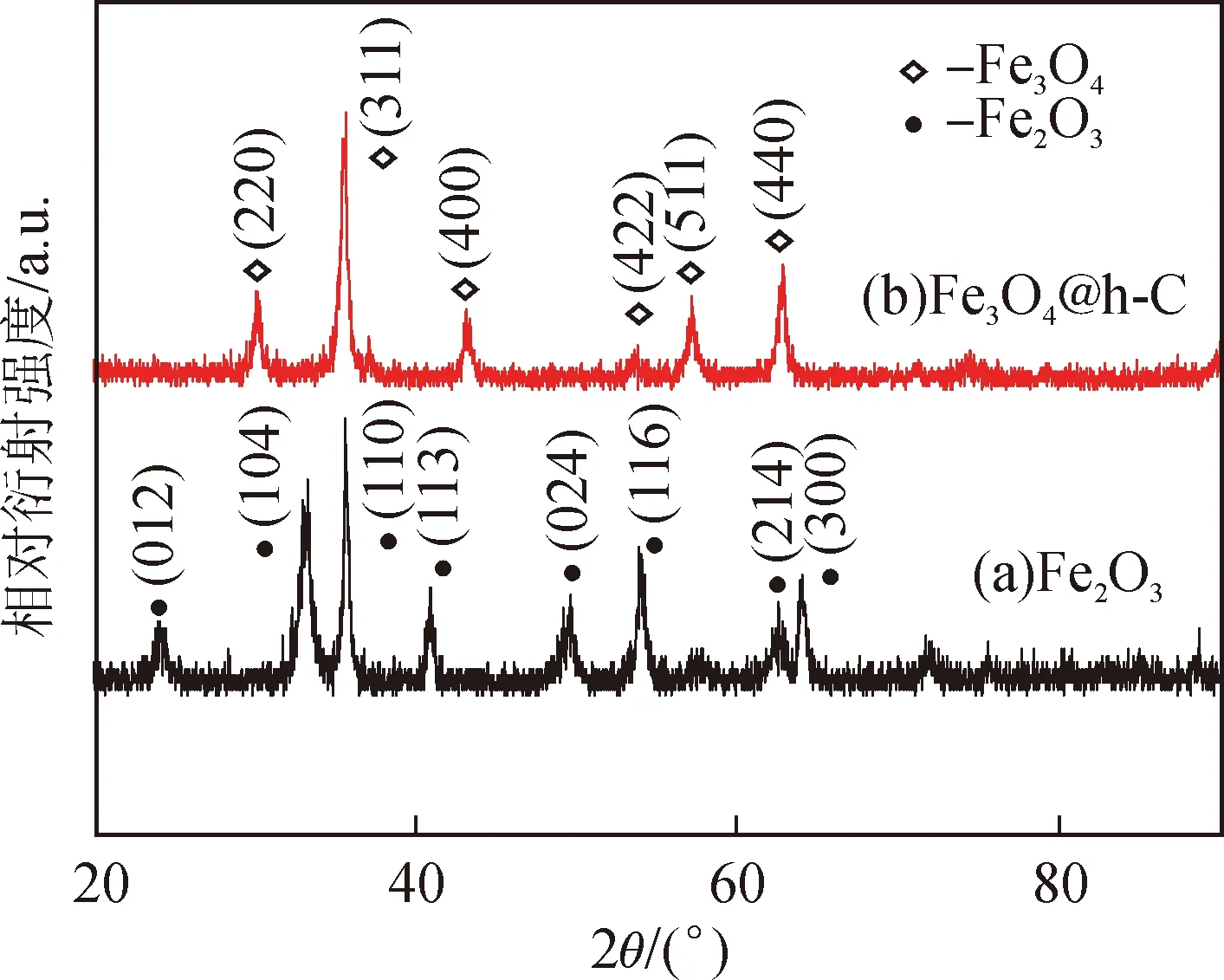
圖2 Fe2O3和Fe3O4@h-C的XRD譜圖Fig.2 XRD patterns of Fe2O3 and Fe3O4@h-C
圖3(a)顯示,Fe3O4@h-C的氮氣吸附脫附等溫曲線是典型的第IV類等溫線,表明產物中存在大量介孔,在P/P0(相對壓力)為0.5~0.9范圍內,曲線上存在一個明顯的滯后環,這是由于其空腔結構對氮氣的吸附造成的[12]。從圖3(b)可以看出,產物的孔尺寸主要在10 nm左右。此外,Fe3O4@h-C的BET比表面積高達119.1m/g,有利于提高其催化活性。
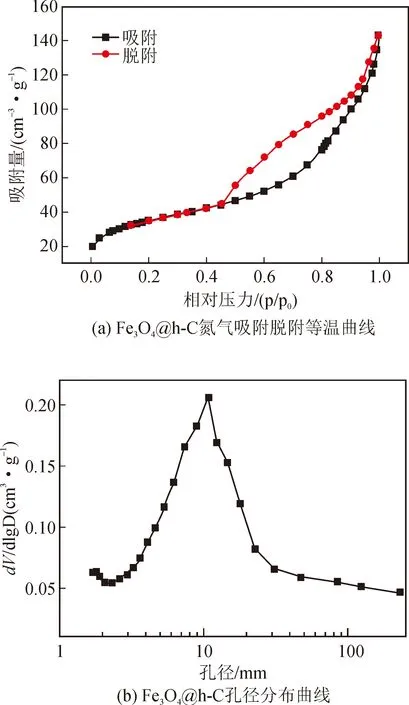
圖3 Fe3O4@h-C的氮氣吸附脫附等溫曲線和孔徑分布曲線Fig.3 Nitrogen adsorption desorption isotherm curve and pore size distribution curve of Fe3O4@h-C
從TEM照片中(圖4(a))可以看出,作為原料的Fe2O3納米棒,其平均長度約為63.0 nm, 寬度為18.7 nm。將該Fe2O3納米棒進行二氧化硅、間苯二酚-甲醛樹脂包覆并熱處理之后,從產物(Fe3O4@SiO2@C)的TEM照片(圖4(b))中可以觀察到樣品具有明顯的核殼結構。其中,Fe3O4納米棒全部位于產物的中間位置,其尺寸約為長43.6 nm,寬14.2 nm,略小于圖4(a)中的Fe2O3,說明在還原過程中發生了體積收縮[13]。此外,產物的殼層厚度約為12.8 nm,但由于碳和二氧化硅具有相似的襯度,二者之間觀察不到明顯的界面[1]。如圖4(c)和(d)所示,經過NaOH刻蝕后,產物都轉換成 “鈴鐺型”結構,說明成功制備了Fe3O4@h-C。Fe3O4@h-C外部的碳殼層厚度約為3.7 nm,并沒有發生變形和塌陷,在有效保護內部Fe3O4的同時,還有利于內外物質的快速通過[1]。值得注意的是,產物內部的Fe3O4偏離了中心位置,說明其在殼層內部具有一定活動能力,有利于催化反應中與有機物充分接觸[14]。
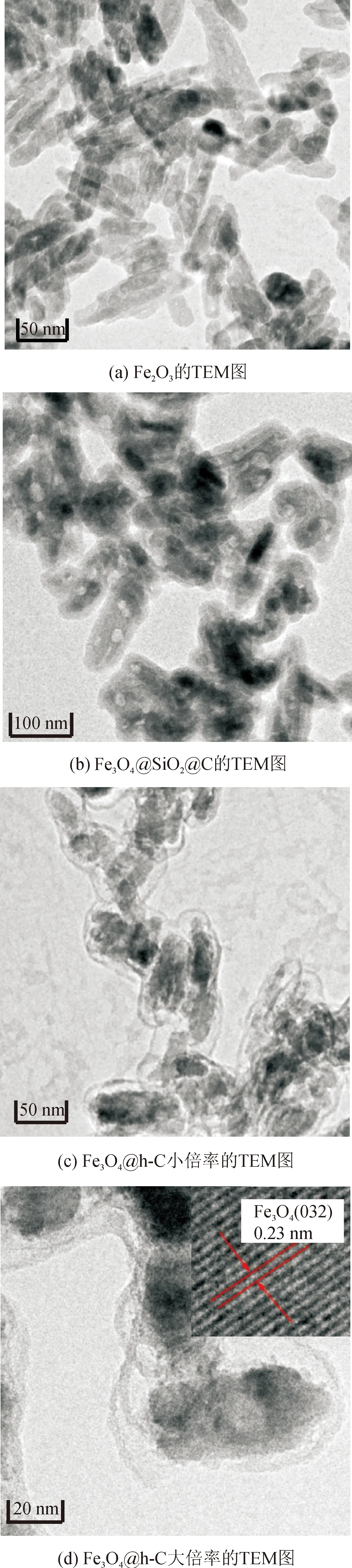
圖4 Fe2O3、Fe3O4@SiO2@C和 Fe3O4@h-C的TEM圖像Fig.4 TEM images of Fe2O3, Fe3O4@SiO2@C and Fe3O4@h-C
圖5是刻蝕前后,樣品(Fe3O4@SiO2@C與Fe3O4@h-C)的紅外特征譜圖。兩種樣品在3000~3700 cm-1處是-OH基團的伸縮振動吸收峰,在1600 cm-1左右的吸收峰來自于苯環骨架的振動,在1400 cm-1左右的吸收峰為-CH2-剪切振動吸收峰,在580 cm-1處的吸收峰來自于Fe-O鍵的伸縮振動[4, 8]。對于刻蝕前的Fe3O4@SiO2@C,其譜圖中位于1095 cm-1處的吸收峰來自于Si-O-Si的反對稱振動[1];對于刻蝕后的Fe3O4@h-C納米棒,其譜圖中歸屬有機基團和Fe3O4的峰依然可見,而歸屬二氧化硅的兩個峰基本消失了,且在1056 cm-1處出現了新的來自于C-O鍵的伸縮振動吸收峰[8]。以上結果表明,刻蝕過后,二氧化硅已被除去,碳化過后的樣品中還有大量的有機基團,這有利于其在水中的分散,有利于催化反應的進行。
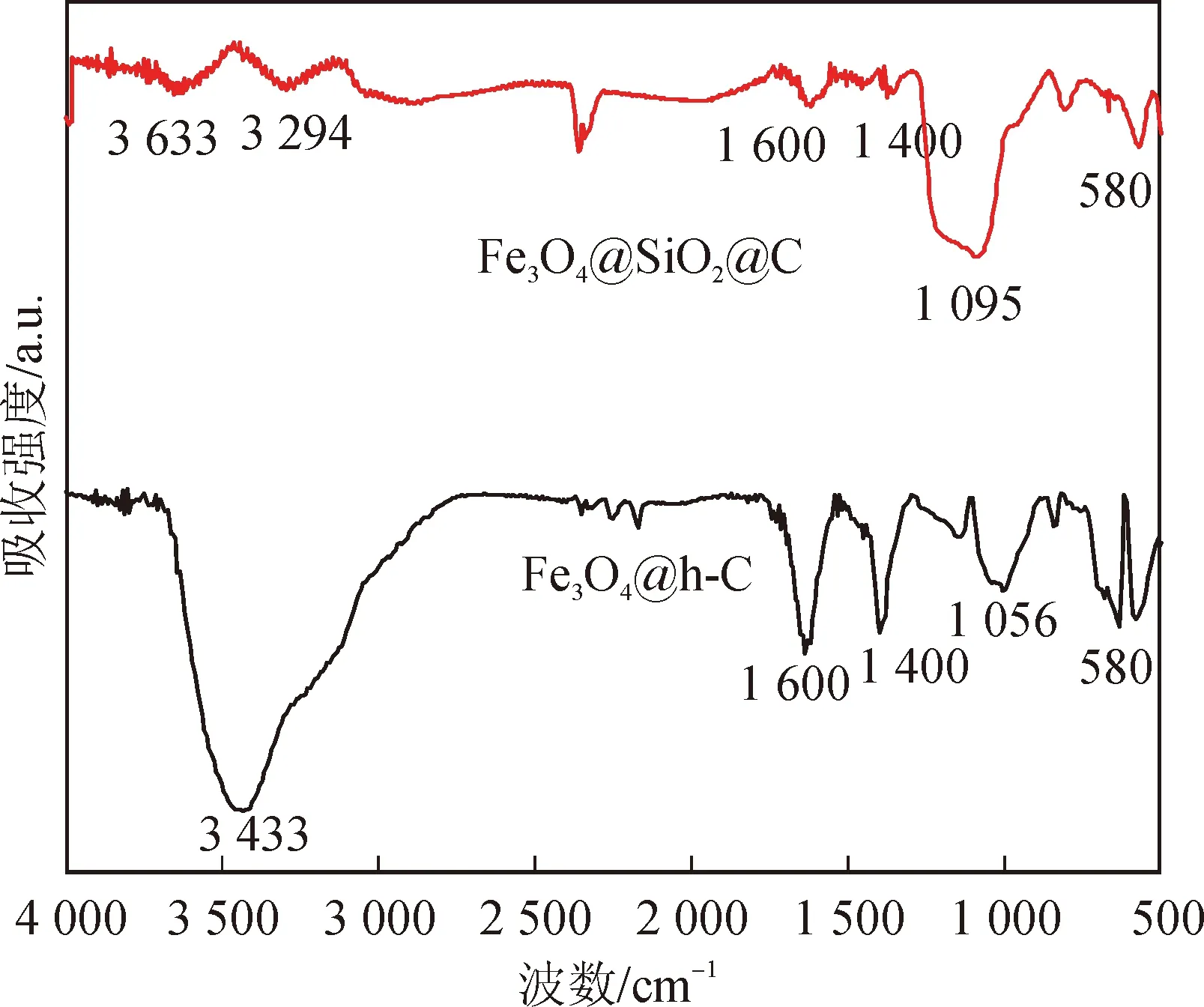
圖5 Fe3O4@SiO2@C和Fe3O4@h-C的傅里葉紅外譜圖Fig.5 FTIR of Fe3O4@SiO2@C and Fe3O4@h-C
為了考察材料結構對催化性能的影響,本研究還分別制備了Fe3O4@C納米棒和純Fe3O4納米棒(圖6(a)和(b))。從相應的XRD數據(圖6(c))可以看出,兩個試樣中都存在著結晶性良好且不含雜質的面心立方Fe3O4晶體,這與Fe3O4@h-C納米棒的結果相一致。值得注意的是,從TEM照片可以看出,Fe3O4@C納米棒的碳殼層厚度約為15.7 nm,要大于Fe3O4@h-C納米棒的碳殼層厚度。
2.2 磁性測定
圖7(a)給出了室溫下Fe3O4@h-C、Fe3O4@C及Fe3O4三種材料的磁滯回線。從圖中可以看出,三種材料的磁滯回線均不通過原點,其矯頑力分別為155、242、129 Oe,呈弱鐵磁性[3]。由文獻報道可知,當Fe3O4的尺寸超過30 nm后,其磁性將由超順磁性向鐵磁性轉變,這與TEM照片的結果是相符的[15]。Fe3O4@h-C、Fe3O4@C及Fe3O4納米棒的比飽和磁化強度(Ms)分別為58.5、65、45.7 emu/g。值得注意的是,由X射線能譜分析(EDS)可知,Fe3O4@h-C和Fe3O4@C中的Fe3O4含量分別為62.6 % 和 49.1 %,但是其Ms均高于純的Fe3O4。這可能是由于聚合物分解會形成獨特的微環境,使產物的磁性增強。不過由文獻可知,以上三種材料的Ms均較高,可以保證樣品具有較強的磁響應能力[2-4]。以Fe3O4@h-C為例(圖7(b)),在一塊兒釹鐵硼永磁體的作用下,數秒鐘即可實現從溶液中的分離。

圖6 Fe3O4@C、Fe3O4的TEM圖像和 Fe3O4@C、Fe3O4的XRD譜圖Fig.6 TEM images of Fe3O4@C, Fe3O4 and XRD patterns of Fe3O4@C, Fe3O4
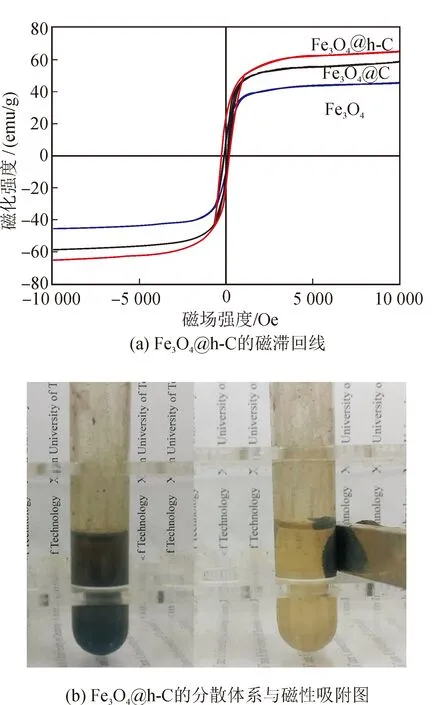
圖7 Fe3O4@h-C的磁滯回線和磁分離效果照片Fig.7 Hysteresis loop and magnetic recycling performance of Fe3O4@h-C
2.3 催化性能測定
由圖8(a)可知,在反應溫度T=50 ℃, pH = 3,攪拌30 min至吸附平衡,加入0.8 mmol/L H2O2的反應條件下,以Fe3O4@h-C為催化劑,加入H2O2僅5 min,亞甲基藍(MB)的褪色率Ct/C0(C0為亞甲基藍溶液初始濃度,Ct為t時刻亞甲基藍溶液濃度)就可以達到95 %;10 min后, MB的褪色率就可達到97.9 %。在同等條件下,純Fe3O4納米棒對于亞甲基藍的褪色率分別為79 %(5 min)和91 %(10 min),低于Fe3O4@h-C。而對于Fe3O4@C, 即使將反應時間延長到30 min,其對MB的降解也僅有21 %。這主要是由于Fe3O4@h-C具有獨特的中空結構,能夠保證溶液中的亞甲基藍分子與內部的Fe3O4顆粒充分接觸,并使得催化劑附近亞甲基藍濃度增加,有利于加快反應速率[1]。相反,對于Fe3O4@C納米棒,較厚的碳殼層阻礙了Fe3O4與溶液中亞甲基藍分子的接觸。而純Fe3O4納米棒在溶液中容易聚集,因此催化性能也低于Fe3O4@h-C。此外,在沒有催化劑的情況下,H2O2自身對MB的褪色率較低;而沒有H2O2加入的情況下,Fe3O4@h-C納米棒對MB基本沒有褪色作用。由此可見,反應是按照Fenton反應機理進行的[8]。
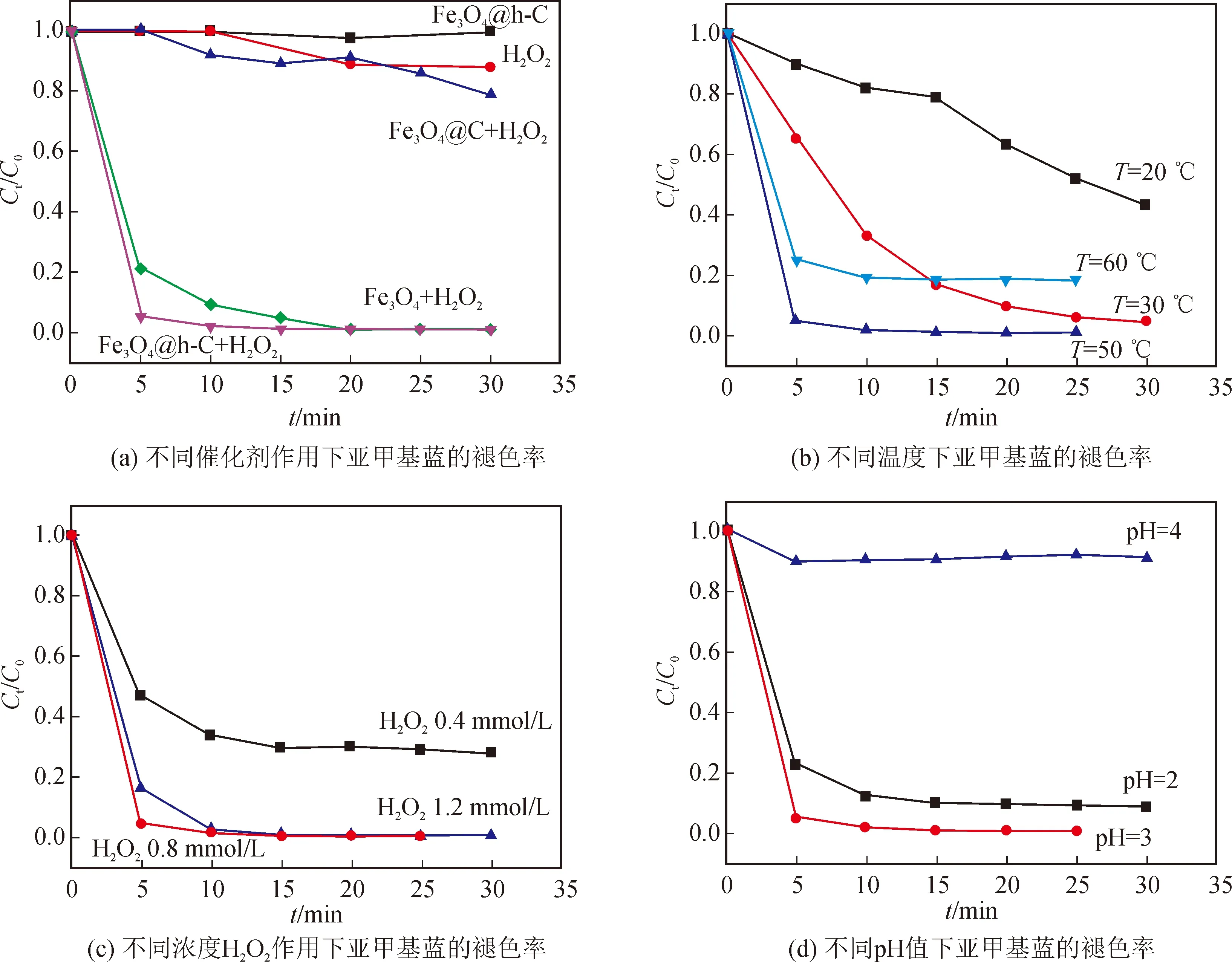
圖8 不同條件下Fe3O4@h-C對亞甲基藍的褪色率Fig.8 Fading rates of MB of Fe3O4@h-C under different conditions
接下來,在保持其它反應條件不變的情況下,分別調節溫度、雙氧水濃度和pH值,探討其對亞甲基藍褪色率的影響。圖8(b)是不同溫度條件下Fe3O4@h-C對亞甲基藍的褪色率,在低于50°C時,MB的褪色速度隨著溫度的升高而加速,而當溫度升高到60°C時,褪色率反而下降,這有可能是由于過高的溫度導致H2O2加速分解,從而降低了反應速率。圖8(c)是不同濃度H2O2條件下,Fe3O4@h-C對亞甲基藍的褪色率,當H2O2濃度較低(0.4和0.8 mmol/L)時,亞甲基藍的降解率隨H2O2濃度的增加而增加;當H2O2濃度大于0.8 mmol/L時,降解率反而有所下降。這是因為在H2O2濃度較低時,H2O2濃度的增加會產生更多的羥基自由基,促進類Fenton反應;而在高H2O2濃度下,過量的H2O2則充當羥基清除劑將羥基自由基轉化為活性較低的自由基,導致降解率下降[12]。圖8(d)是不同pH值條件下,Fe3O4@h-C對亞甲基藍的褪色率,可以發現,反應的最佳pH值為3。當pH值增加到4及以上時,亞甲基藍的降解率急劇下降,這有可能是因為生成了無活性的離子(FeO2+);當pH值降低為2時,亞鐵離子會形成活性較低的鐵絡合物,并且高濃度的H+與H2O2形成穩定的[H3O2]+,減少了羥基自由基的產生,從而降低了亞甲基藍的降解率[12]。圖9是Fe3O4@h-C納米棒的循環催化實驗結果。由圖可知,Fe3O4@h-C納米棒表現出良好的穩定性,在循環使用四次后,MB的降解率依然可以達到92.8 %,表現出了良好的重復使用性,易于回收再利用。
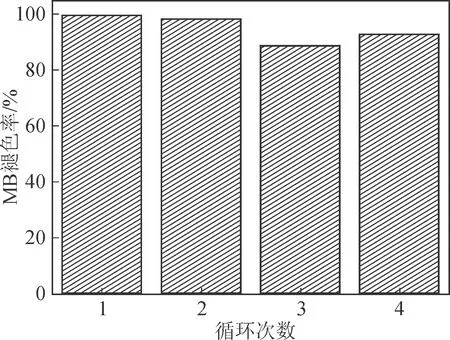
圖9 Fe3O4@h-C的循環催化反應結果Fig.9 Recycling results of the Fe3O4@h-C nanorod catalyst
3 結 論
本文以商用Fe2O3納米棒為起始原料,通過溶膠凝膠法和氫氣還原的方法制備了Fe3O4@h-C納米棒。產物具備磁分離能力。在催化H2O2降解亞甲基藍的反應中,Fe3O4@h-C納米棒展現出良好的催化性能和重復使用性,顯示出一定的實際應用價值。通過對比發現,中空結構不但有利于阻止Fe3O4的聚集,還能保證其與有機污染物充分接觸,對Fe3O4納米棒催化性能的提高是十分有益的。
