密度泛函理論研究吲哚并咔唑同分異構體結構,芳香性和光譜性質
2019-04-28 08:52:38郭雅晶周瑤瑤李秀燕
原子與分子物理學報 2019年5期
郭雅晶, 周瑤瑤, 李秀燕
(1. 太原師范學院物理系,晉中 030619; 2. 太原理工大學物理與光電工程學院,太原 030024)
1 引 言
吲哚并咔唑化合物存在于許多純天然產品和人工合成材料中[1]. 由于吲哚環的稠合是在不同的位置上并且以不同的方式稠合,所以這類化合物具有五種基本骨架結構(即五種同分異構體):吲哚并[2,3-a]咔唑,吲哚并[2,3-b]咔唑,吲哚并[2,3-c]咔唑,吲哚并[3,2-a]咔唑,吲哚并[3,2-b]咔唑[1]. 這五個分子通過共享C-C鍵和不同方位及方位角上的N個原子來得以區分,并且這些分子已被很多研究者通過實驗來測試[2]. 1977年,歐姆拉研究小組捕獲了第一個吲哚[2,3-a]咔唑生物堿,該生物堿被命名為星形鏈霉菌屬的星形孢菌素[2]. 在后來的研究中,許多研究人員從土壤和海洋微生物中分離出大量吲哚卡唑化合物[3,4]. 到目前為止,這種化合物已被分離出來的數量已超過130種[5]. 吲哚并[2,3-a]咔唑具有大環分子的基本結構,還具有陰離子識別作用,因此可作為化學傳感器使用[6]. 此外,由于吲哚并[2,3-a]咔唑具有豐富的電效應,而且在芳香族化合物聚集的超分子化學中具有自組裝的特性[7]. 吲哚并[3,2-b]咔唑具有相對較寬的帶隙和較低的最高占據分子軌道(HOMO),這使得它在空氣中具有比戊烯較強的氧化穩定性[8,9],從而在有空氣的環境條件下實現了低成本的制造. 此外,吲哚并[3,2-b]咔唑共聚物被報道為潛在的光伏材料[10,11].
這些化合物通常具有較寬的帶隙和較低的高占據分子軌道(HOMO). 這些分子的共軛平面結構有助于載流子傳輸,再加之其大平面剛性結構使這些化合物具有良好的熱穩定性. 吲哚并咔唑化合物由于其良好的發光性能和潛在的應用前景被視為研究熱點;隨著其生物活性逐漸受到研究者關注,這些化合物作為分子材料在材料應用研究方面也逐漸成為熱點,并顯示出巨大的潛在應用前景[12-14]. 基于這類分子骨架的特殊性,通過應用理論計算方法研究了五種吲哚并咔唑異構體的電子結構、芳香性和光譜性質. 據研究所知,吲哚并咔唑異構體的芳香性和重組能尚未得到研究. 下文計算結果表明,這五個同分異構體適用于P型傳輸材料,每個分子的三個苯環均具有共軛效應.
2 計算細節
所有計算均使用高斯09軟件包進行[15]. 利用Becker的混合三參數泛函在Lee, Yang和Parr相關泛函(B3LYP)[16]水平上與6-31G(d,p)基組的密度泛函理論(DFT)研究了吲哚并[2,3-a]咔唑、吲哚并[2,3-b]咔唑、吲哚并[2,3-c]咔唑、吲哚并[3,2-a]咔唑和吲哚并[3,2-b]咔唑這五種分子的結構. 還考慮了不同的自旋多重性和初始結構,為了證實吲哚并咔唑異構體團簇結構的穩定性,還分析了振動頻率. 諧波振動頻率分析也在相同的理論水平上進行,以找出勢能面上的最小值. 這里考慮的所有分子都具有零數量的虛頻率(NIMAG=0). 分子軌道(MO)圖像通過GAUSSVIEW 5.0[17]軟件生成. 此外,還研究了電離勢(垂直和絕熱)和電子脫離能(垂直和絕熱). 團簇芳香性通過離域分子軌道、非獨立核化學位移(NICSs)[18]、共振能量等來評估. 為了表示σ或π芳香性的性質,在B3LYP/6-31G(d,p)基組水平上,使用GIAO[19]計算了環中心或籠中心[NICS(0)]和環平面上1?[NICS(1)]處的NICS值. 共振能量使用B3LYP方法和6-31G(d,p)基組來細化. 通過B3LYP/6-31G(d,p)計算,獲得了吲哚并[2,3-a]咔唑、吲哚并[2,3-b]咔唑、吲哚并[2,3-c]咔唑、吲哚并[3,2-a]咔唑和吲哚并[3,2-b]咔唑的最高占據軌道(HOMO)和最低未被占據軌道(LUMO)能量間隙.
為了研究溶劑對所研究復合物的選定性質的影響,因此選擇極化連續介質模型(PCM)中的自洽反應場(SCRF)進行理論計算[20,21]. 因此,在B3LYP/6-31G(d,p)[22]理論水平上,采用含時密度泛函理論(TD-DFT)方法進行了吸收光譜和發射光譜的計算,并通過使用可極化連續體模型(PCM)方法,在二氯甲烷溶劑中求解了20個激發態的光譜.
3 結果和討論
3.1 幾何結構
構建了吲哚并[2,3-a]咔唑、吲哚并[2,3-b]咔唑、吲哚并[2,3-c]咔唑、吲哚并[3,2-a]咔唑和吲哚并[3,2-b]咔唑的多種可能結構,此外還考慮了這些分子的不同自旋態,最后經過B3LYP/6-31g(d,p)優化以及計算頻率,確定了最低能量穩定結構. 這五個同分異構體分子的最低能量穩定結構如圖1所示,并且在同基組水平下計算了吲哚并咔唑異構體的主要鍵長、鍵角和扭轉角,見表1. 結果表明,吲哚并[3,2-a]咔唑的C-N鍵長和C-C鍵長與Zhao G[23]的結果一致;然而,吲哚并[2,3-a]咔唑的C-N鍵長度分別為1.388和1.390?,此結果略長于Zhao G(分別為1.38和1.39?),這是由于兩者所選取的高斯軟件版本不同.

圖1 吲哚并咔唑同分異構體結構Fig. 1 Structures of indolocarbazole isomers.
表1 吲哚并咔唑同分異構體的主要鍵長(?),鍵角(°)和扭轉角(°)
Table 1 The main bond lengths(?), bond angles(°) and torsion angles(°) of indolocarbazole isomers
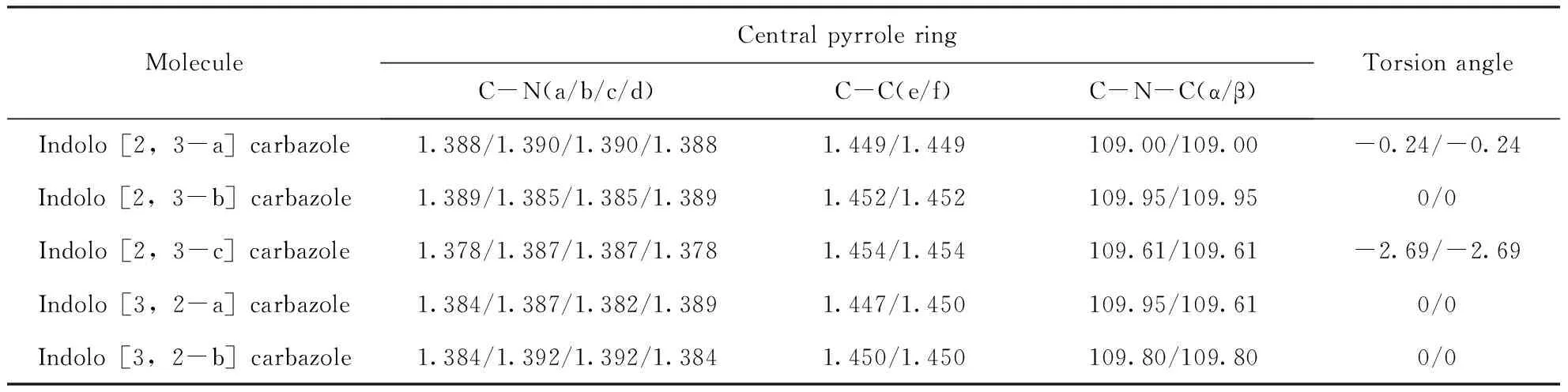
MoleculeCentral pyrrole ringC-N(a/b/c/d)C-C(e/f)C-N-C(α/β)Torsion angleIndolo[2,3-a]carbazole1.388/1.390/1.390/1.3881.449/1.449109.00/109.00-0.24/-0.24Indolo[2,3-b]carbazole1.389/1.385/1.385/1.3891.452/1.452109.95/109.950/0Indolo[2,3-c]carbazole1.378/1.387/1.387/1.3781.454/1.454109.61/109.61-2.69/-2.69Indolo[3,2-a]carbazole1.384/1.387/1.382/1.3891.447/1.450109.95/109.610/0Indolo[3,2-b]carbazole1.384/1.392/1.392/1.3841.450/1.450109.80/109.800/0
表2給出了B3LYP/6-31g(d,p)計算所得的基態參數,即基態對稱性、最低頻率、結合能、平均結合能、溫度、壓力、零點能、最高占據能HOMO、最低未被占據能LUMO和能隙. 吲哚并咔唑異構體的點群(基態對稱性)參數分別為C2、C2V、C2、CS和C2h,使用該水平基組計算的最低頻率都是正數,這意味著它們位于勢能面的局部最小值. 基態結構下結合能與平均結合能的計算結果均為負數,表2中的基態參數進一步說明B3LYP/6-31g(d,p)下優化后的吲哚并咔唑異構體是基態穩定結構. 該計算中設定優化環境溫度為298.15 K,壓力為1 atm,此外,能量間隙是指最高占據軌道(HOMO)能量值與最低未被占據軌道(LUMO)能量值之差的絕對值. HOMO、LUMO和能隙的結果與Govindaswamy Ranganathan Ramkumaar[24,25]的結果一致. 這更進一步說明,所選取的理論計算方法及基組水平是合理的. 吲哚并[3,2-b]咔唑的HOMO、LUMO和能隙值分別為-4.932、-1.015和3.917 eV,與其他吲哚并咔唑異構體相比其能隙最小,這是由于它的苯基和核心組間HOMO (LUMO)的能量差異越大,導致干擾相互作用越小,因此吲哚并[3,2-b]咔唑中苯基對HOMO (LUMO)的貢獻越小.

表2 吲哚并咔唑同分異構體基態參數
3.2 重組能
圖2列出了所有前線分子軌道(圖中橫坐標下的1,2,3,4,5分別對應吲哚并[2,3-a]咔唑,吲哚并[2,3-b]咔唑,吲哚并[2,3-c]咔唑,吲哚并[3,2-a]咔唑和吲哚并[3,2-b]咔唑分子). 如圖2所示,五個分子的HOMO和LUMO能級π軌道特征以及離域的程度均在所有異構體的原子核,與此同時HOMO軌道主要執行π鍵軌道而LUMO軌道主要執行π*反鍵軌道. 除了吲哚并[3,2-a]咔唑外,其他異構體的N原子上的p軌道不會與苯環平面形式有效p-π共軛,與此同時在LUMO軌道上N原子的p軌道幾乎沒有貢獻. 吲哚并[3,2-a]咔唑的N原子之一是在HOMO軌道上形成有效的p-π共軛,盡管來自N原子的p軌道組成比例在LUMO軌道上降低,但仍然高于其他四個分子.

圖2 吲哚并咔唑同分異構體的最高占據軌道和最低未被占據軌道能級Fig.2 The HOMO and LUMO orbital energy levels of indolocarbazole isomers.
表3給出了這五個同分異構體的電離勢(IP,eV)、電子親和能(EA,eV)、空穴/電子提取勢(HEP/EEP,eV)和空穴/電子重組能量(λh/λe,eV). 從表2和表3可以看出,共軛鍵的電離勢和電子親和能的變化與HOMO和LUMO能級的變化一致,相同共軛鍵的化合物的HOMO能級顯著降低,同時LUMO能級幾乎不變;反映在電離勢和電子親和能上,電離勢顯著增加,電子親和能基本相等. 另一方面,如圖2中所示,吲哚并咔唑異構體的HOMO能級大小順序為5>3>2>4>1,并伴隨有電離勢隨其順序逐漸增加;異構體的LUMO能級大小順序為4>2>1>3>5,電子親和能值隨其順序逐漸增加. 從表3中可以看出,這些分子團簇的空穴/電子重組能量值λh和λe大小順序為:4>2>1>3>5. 吲哚并[3,2-b]咔唑的空穴/電子重組能值最小,說明吲哚并[3,2-b]咔唑的空穴遷移率和電子遷移率在吲哚并咔唑異構體中最好. 此外,吲哚并[3,2-b]咔唑的空穴重組和電子重組能更接近,所以空穴遷移率相當于電子遷移率,有利于載體平衡傳輸和傳輸效率. 載流子的傳輸與前線分子軌道的分布密切相關,通常LUMO (HOMO)能級的離域化程度越大,越有利于電子(空穴)的傳輸[26]. 劉蓮梅科研小組[26]確認p型輸運材料IPA在5.680~6.786 eV范圍內,n型輸運材料EAA在2.411~3.141 eV范圍內,IPA和EAA材料的雙極性傳遞應在5.905~7.026 eV和2.797~3.479 eV范圍內,從表3可以看出,這五個分子均屬于p型輸運材料.
表3 吲哚并咔唑同分異構體的電離勢(IP, eV)、電子親和能(EA, eV)、空穴/電子提取勢(HEP/EEP, eV)和空穴/電子重組能量(λh/λe, eV)
Table 3 Ionization potentials(IP, eV), electronic affinities(EA, eV), hole/electron extraction potentials(HEP/EEP, eV) and hole/electron reorganization energies(λh/λe, eV) of indolocarbazole isomers
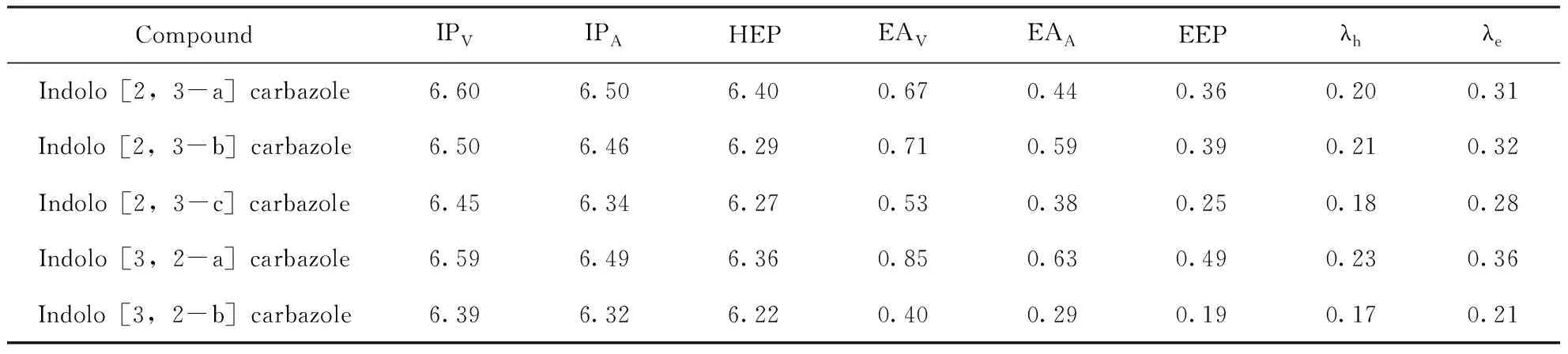
CompoundIPVIPAHEPEAVEAAEEPλhλeIndolo[2,3-a]carbazole6.606.506.400.670.440.360.200.31Indolo[2,3-b]carbazole6.506.466.290.710.590.390.210.32Indolo[2,3-c]carbazole6.456.346.270.530.380.250.180.28Indolo[3,2-a]carbazole6.596.496.360.850.630.490.230.36Indolo[3,2-b]carbazole6.396.326.220.400.290.190.170.21
3.3 芳香性
采用GIAO-B3LYP/6-31g(d, p)法計算吲哚并咔唑異構體的核獨立化學位移值(NICS)來表征這些分子的芳香性. 將當前NICS的參考點設置在兩個位置,如表4中,NICS(0.00nm)位于吲哚并咔唑異構體每個環的幾何中心,NICS(0.10nm)位于幾何中心垂直距離0.10nm處;分別在每個位置放置一個鬼(ghost)原子,并計算相應的NICS值. 結合圖1與表4得出,所有吲哚并咔唑異構體中的每個環均具有芳香性. 如表4所示,隨著鬼原子垂直距離的增大,所有分子的環2、環3、環4的芳香性減小;相反,環1和環5的芳香性隨著鬼原子垂直距離的增加而增加. 對于芳香性體系,通常屏蔽值是正的,而NICS是負的,主要是因為共軛環引起的外部磁場對環電流感應磁場有一定程度的抵消(屏蔽)作用. NICS(0.10 nm)主要測量的是π芳香性. 由以上結論可知,每個分子的三個苯環都具有共軛效應.
3.4 吸收和發射譜
用高斯曲線擬合的吲哚并咔唑同分異構體的吸收和發射光譜如圖3和圖4所示. 這五個分子的吸收光譜曲線特征(圖3)都具有兩個顯著的吸收峰和一個較差的吸收峰. 最低能量的吸收峰(>290 nm)主要是由于每個環上的每個分子的π-π*鍵過渡引起的,而較高能量的吸收峰(200-290 nm)主要是由于N原子雜環和苯環之間的π-π*鍵過渡引起的. 吲哚并咔唑異構體的垂直激發能和擬合的吸收峰列于表5中,結果表明,吲哚并[2,3-a]咔唑和吲哚并[3,2-a]咔唑的激發能與實驗吸收波長吻合較好[27],從實驗中分子體系的中心苯環對甲基取代基的影響來看,存在著細微的差異. 上述結果表明,基于這些分子的仿真方法能夠得到較好的結果.
雖然同分異構體的吸收光譜差異較小,但發射光譜(圖4)結果有明顯差異. 表6列出了吲哚并[2,3-a]咔唑和吲哚并[3,2-a]咔唑的同分異構體轉移特征、理論計算值及相關實驗值[27]. 將表5和表6對比可得:吲哚并[2,3-a]咔唑的最低發射能主要來自電子LUMO→HOMO躍遷,而其最低吸收能主要來自電子LUMO→HOMO-1躍遷;按照對稱性,吲哚并[2,3-a]咔唑中來自電子LUMO→HOMO躍遷產生的最低發射能主要對應于其電子LUMO→HOMO-1躍遷產生的最低吸收能量,剩余其他分子的電子躍遷產生的最低發射能也來自電子躍遷產生的最低吸收能. 比較振子強度發現,三個近似線性分子(吲哚并[2,3-a]咔唑,吲哚并[2,3-b]咔唑和吲哚并[3,2-b]咔唑)的電荷轉移躍遷振子強度更大,吲哚并[2,3-c]咔唑和吲哚并[3,2-a]咔唑可以由于其構型特征而保持高能發射. 比較結果表明,吲哚并[2,3-b]咔唑在這些分子中的振子強度最大.
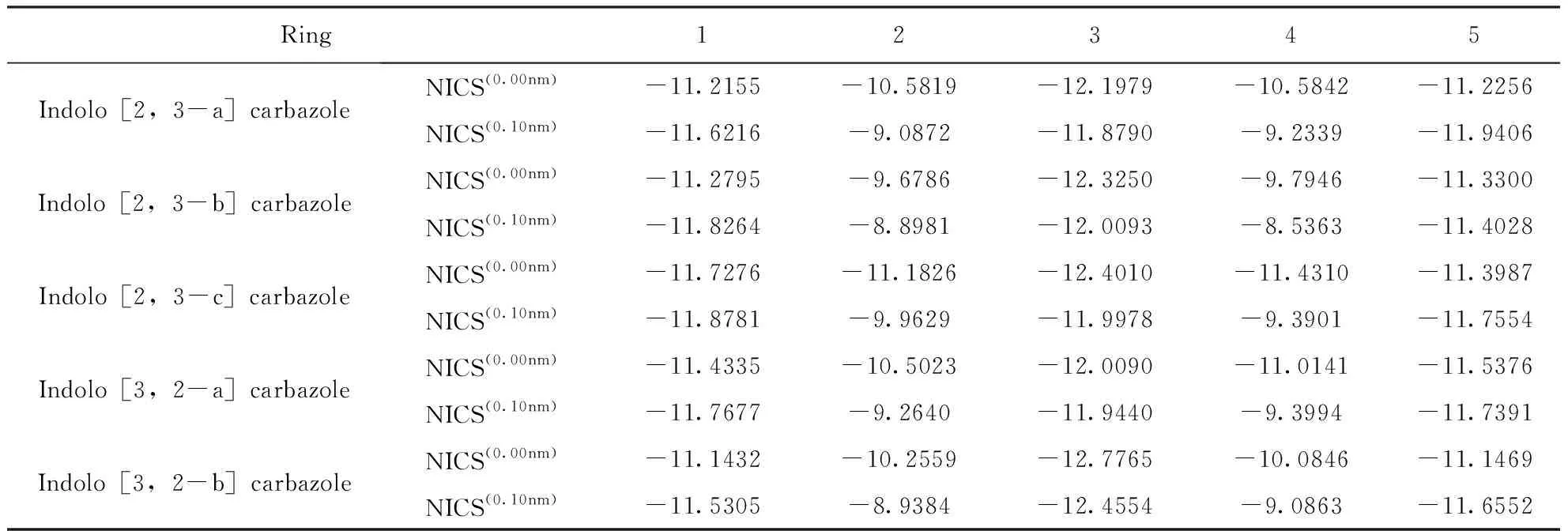
表4 吲哚并咔唑同分異構體基態下的核獨立化學位移值NICS(ppm)
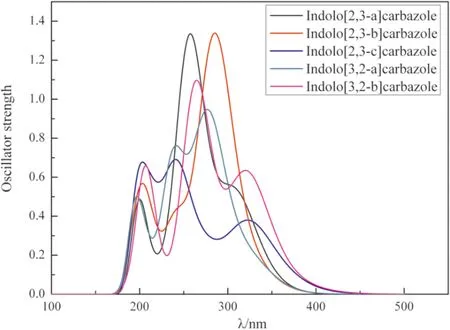
圖3 吲哚并咔唑同分異構體在CH2Cl2溶劑下的吸收光譜Fig. 3 Absorption spectra of indolocarbazole isomers in CH2Cl2 solution.
4 結 論
基于密度泛函理論(B3LYP)在6-31g(d,p)水平上,研究了優化后的性能(基態結構參數、結合能、能隙、芳香性、重組能). 結果表明,吲哚并咔唑異構體(吲哚并[2,3-a]咔唑,吲哚并[2,3-b]咔唑,吲哚并[2,3-c]咔唑,吲哚并[3,2-a]咔唑和吲哚并[3,2-b]咔唑)基態結構對稱性分別為C2、C2V、C2、CS和C2h.
表5 用TD-DFT計算得到吲哚并咔唑異構體在CH2Cl2溶劑中的吸收特性
Table 5 Absorption properties of indolocarbazole isomers in CH2Cl2solution obtained with the TD-DFT calculations

IsomerTransitionEver/nmOscillatorstrengthλexp/nm[27]Indolo[2,3-a]carbazole1A→1BHOMO-1→LUMO(89%)308.040.59043231A→1BHOMO→LUMO+2(84%)256.151.34542591A→1BHOMO-3→LUMO+1(46%)HOMO-2→LUMO+3(50%)199.680.4957Indolo[2,3-b]carbazole1A1→1B2HOMO→LUMO+1(73%)285.521.35631A1→1B2HOMO-1→LUMO+2(82%)243.670.45541A1→1B2HOMO-2→LUMO+2(47%) HOMO→LUMO+5(43%)202.780.5845Indolo[2,3-c]carbazole1A→1BHOMO-1→LUMO(95%)323.420.39011A→1BHOMO→LUMO+2(94%)246.950.70091A→1AHOMO-2→LUMO+2(57%)198.400.6997Indolo[3,2-a]carbazole1A’→1A’HOMO-1→LUMO+1(68%)277.540.95382821A’→1A’HOMO-3→LUMO(29%)HOMO-2→LUMO+1(32%)247.140.75301A’→1A’HOMO-2→LUMO+3(33%)HOMO-1→LUMO+5(43%)194.380.5018Indolo[3,2-b]carbazole1BU→1BUHOMO-1→LUMO(94%)321.500.65381BU→1BUHOMO→LUMO+2(93%)264.831.10481BU→1BUHOMO-1→LUMO+4(58%)208.960.6807

圖4 吲哚并咔唑同分異構體在CH2Cl2溶劑下的發射光譜Fig. 4 Emission spectra of indolocarbazole isomers in CH2Cl2 solution.
表6 用TD-DFT計算得到吲哚并咔唑異構體在CH2Cl2溶劑中的發射特性
Table 6 Emission properties of indolocarbazole isomers in CH2Cl2solution according to the TD-DFT calculations

IsomerTransitionEver/nmOscillatorstrengthλexp/nm[27]Indolo[2,3-a]carbazole1A→1ALUMO→HOMO(100%)365.900.09073911B→1ALUMO→HOMO-1(93%)323.250.87221B→1ALUMO+1→HOMO(89%)289.030.3871Indolo[2,3-b]carbazole1A1→1A1LUMO→HOMO(100%)351.260.03901B2→1A1LUMO→HOMO-1(85%)335.950.33691B2→1A1LUMO+1→HOMO(84%)300.521.7595Indolo[2,3-c]carbazole1A→1ALUMO→HOMO(100%)395.210.15041B→1ALUMO→HOMO-1(100%)335.910.58031B→1ALUMO+1→HOMO(100%)301.090.2030Indolo[3,2-a]carbazole1A’→1A’LUMO→HOMO(95%)348.550.38213701A’→1A’LUMO→HOMO-1(90%)320.500.13423551A’→1A’LUMO+1→HOMO(88%)294.770.3499Indolo[3,2-b]carbazole1BU→1BULUMO→HOMO(100%)392.420.09141BU→1BULUMO→HOMO-1(98%)335.971.1132
這五個分子均是p型輸運材料. 由以上結論可知,每個分子的三個苯環具有共軛效應. 通過與實驗結果的比較,得到了更精確的理論計算結果,與實驗結果吻合較好. 由于幾何結構的不同,五種吲哚并咔唑異構體在性質上存在明顯差異. 線性分子(吲哚并[2,3-a]咔唑,吲哚并[2,3-b]咔唑和吲哚并[3,2-b]咔唑)有助于提高環間電荷轉移躍遷的振子強度.
