Novel frameshift mutation causes early termination of the thyroxinebinding globulin protein and complete thyroxine-binding globulin deficiency in a Chinese family:A case report
2019-04-22 06:28:14PingPingDangWeiWeiXiaoZhongYanShanYueXiRanRanWangXiaoHuiYuWeiPingTengXiaoChunTeng
World Journal of Clinical Cases 2019年22期
Ping-Ping Dang, Wei-Wei Xiao, Zhong-Yan Shan, Yue Xi, Ran-Ran Wang, Xiao-Hui Yu, Wei-Ping Teng,Xiao-Chun Teng
Ping-Ping Dang, Wei-Wei Xiao, Zhong-Yan Shan, Ran-Ran Wang, Xiao-Hui Yu, Wei-Ping Teng,Xiao-Chun Teng, Department of Endocrinology and Metabolism, Institute of Endocrinology,Liaoning Provincial Key Laboratory of Endocrine Diseases, The First Hospital of China Medical University, Shenyang 110001, Liaoning Province, China
Yue Xi, Department of Endocrinology and Metabolism, The Third Affiliated Hospital of Jinzhou Medical University, Jinzhou 121000, Liaoning Province, China
Abstract
Key words: Thyroxine-binding globulin; Complete thyroxine-binding globulin deficiency;Partial thyroxine-binding globulin deficiency; Gene polymorphism; Case report
INTRODUCTION
Thyroxine-binding globulin (TBG) is the main thyroid hormone transport protein in humans, carrying approximately 75% of the total thyroxine (TT4) and 70% of the total triiodothyronine (TT3) present in serum[1-3].The humanTBGgene belongs to the serpin family of genes and is located on the long arm of the X-chromosome (Xq22.2)[4,5].Abnormalities in TBG are caused by mutations in theTBGgene, and demonstrate an X-linked pattern of inheritance[6-8].Based on the serum levels of TBG in hemizygotes expressing only the mutant allele, TBG defects are classified as complete TBG deficiency (TBG-CD), partial TBG deficiency (TBG-PD), and TBG excess (TBG-E)[1].
To date, 28TBGmutations that cause TBG-CD have been identified:7 intron region mutations, 20 exon mutations, and 1 mutation involving both an intron and an exon.These 28 mutations include 14 single nucleotide substitutions, 12 nucleotide deletions,1 deletion-insertion, and 1 single nucleotide insertion.Additionally, 19TBGgene mutations result in TBG-PD; all of them are single nucleotide substitutions, with 17 in exon regions, 1 in an intron, and 1 in the downstream enhancer region of theTBGgene.Furthermore, 3 single nucleotide substitutions inTBGhave been identified as gene polymorphisms that do not cause changes in TBG levels[9-14].As yet, no large insertional mutations that associate with TBG-CD have been reported inTBG.
Here, we report a novelTBGmutation in exon1, c.381_382 ins TTGCAGATAGGAAATGCCC (p.Phe135Alafs*21), which was associated with TBG-CD in a Chinese family.This 19-nucleotide insertion in exon 1 resulted in a frameshift and a premature stop codon at position 155 of the protein coding sequence; the mutation is termed TBG-CDC.The proband and his brother are hemizygous for the mutation, and manifested the TBG-CD phenotype.The proband’s mother is heterozygous for this mutation, but displayed the same TBG-CD phenotype as her affected sons.The proband’s father has a single nucleotide substitution in exon 3, c.909G>T(p.Leu303Phe), which is known asTBG-Poly (L283F).The proband’s father had low TT4and TT3, but a normal amount of thyrotropin (TSH), and his serum TBG level was between normal and affected hemizygous, which is inconsistent with previous reports that this polymorphism causes no changes in TBG levels.
CASE PRESENTATION
Chief complaints
A 46-year-old Chinese man had a health examination at a local hospital.He had normal free thyroxine (FT4), free triiodothyronine (FT3), and TSH, but low TT4and TT3.The patient was then referred to our hospital for additional diagnosis.
History of present illness
The patient underwent physical examination a month ago and abnormal thyroid function was found.
History of past illness
The patient had a free previous medical history.
Physical examination
The patient’s thyroid gland was normal, without nodules, as measured by thyroid gland ultrasound.The patient denied symptoms of weakness, drowsiness, and intolerance to cold.
Laboratory examinations
Upon re-evaluation of the patient’s thyroid function, we detected low serum TT4and TT3levels, normal FT4, FT3, and TSH levels, and undetectable serum TBG (the lower limit of detection of the assay was 3.5 μg/mL; normal range:14–31 μg/mL).
Further diagnostic work-up
To find out if other people in the proband's family have similar performance, blood samples were obtained from the patient’s family members, including his paternal grandfather, parents, younger brother, and nephew.To identify the prevalence of theTBG-Poly (L283F) variant in Chinese men, serum and whole blood samples were obtained from 117 unrelated Han Chinese men from northeastern China.
Serum TT4, TT3, FT3, FT4, TSH, thyroglobulin antibodies (TgAb), and thyroperoxidase antibodies (TPOAb) were measured using electrochemiluminescence immunoassays performed on a Cobas Elesys 601 instrument.TT4,TT3, FT3, FT4, TSH, TgAb, TPOAb were analyzed using the competition principle, and TSH using the sandwich principle (Catalog number:26047103 for TSH, 23816601 for TT3, 25785701 for FT3, 24807703 for TT4, 24682503 for FT4, 22084502 for TPOAb, and 23015003 for TgAb, Roche Diagnostics GmbH, Mannheim, Germany).Serum TBG and thyroglobulin (Tg) were measured using a solid–phase, competitive chemiluminescent enzyme immunoassay on an Immulite 2000 Xpi instrument(Catalog number:L2KTB2 for TBG and L2KTY2 for Tg, Siemens Healthcare Diagnostics Products limited, United Kingdom).Genomic DNA was isolated from peripheral blood using a DNA Kit (TIANGEN, Beijing, China), and targeted sequences were amplified by PCR.Gene regions including exons 1–4 of theTBGgene and intron-exon boundaries were sequenced (3730XL; Applied Biosystems, Carlsbad,California).Quantitative PCR high-resolution melting curve analysis was used to screen theTBG-Poly (L283F) variant among 117 Chinese men; the results were verified by direct DNA sequencing.The primers used in PCR amplification and sequencing are shown in Table 1.
The pedigrees and results of the thyroid function tests (TFTs) of the family members are shown in Figure 1.The proband (III-3), his brother (III-2), and his mother (II-1) had low serum TT4and TT3levels but normal TSH concentrations, and serum TBG was undetectable, which is characteristic of TBG-CD.The proband’s father (II-2) had low TT4and TT3, but normal TSH; his serum TBG level was between normal and affected hemizygous (Figure 1), which indicated TBG-PD.The proband’s grandfather (I-2) and nephew (IV-1) had normal TFTs.All family members had normal thyroglobulin (Tg), TgAb, and TPOAb levels.
Two mutations in theTBGgene were identified in this Chinese family.One, a novel mutation, was identified in the index III-3, III-2, and II-1.This mutation is a 19-nucleotide insertion, occurring between cDNA positions 381 and 382(c.381_382insTTGCAGATAGGAAATGCCC) in exon 1.This mutation changes the phenylalanine at codon 135 to alanine, following which there are 19 amino acids and then an early termination codon at position 155, leading to premature termination of TBG (Figure 2A).This mutation results in a truncated protein containing only the first 134 amino acids; in comparison, the wild-type TBG protein (TBG-C) is 395 amino acids long, excluding the 20 amino acid signal peptide.As expected, the III-3 and III-2 are hemizygous for theTBGmutation, while II-1 is heterozygous, demonstrating that the mutation follows a pattern of X-linked inheritance.
The otherTBGmutation is a single nucleotide substitution (TTG→TTT) in codon303 in exon 3 (p.Leu303Phe), which was identified in the II-2 (Figure 2B).This variant was previously known asTBGpolymorphism,TBG-Poly (L283F).The II-2 carried theTBG-Poly (L283F) polymorphism and presented with TBG-PD.He had low TT4and TT3levels, with his serum TBG levels falling between normal and affected hemizygous levels (Figure 1).
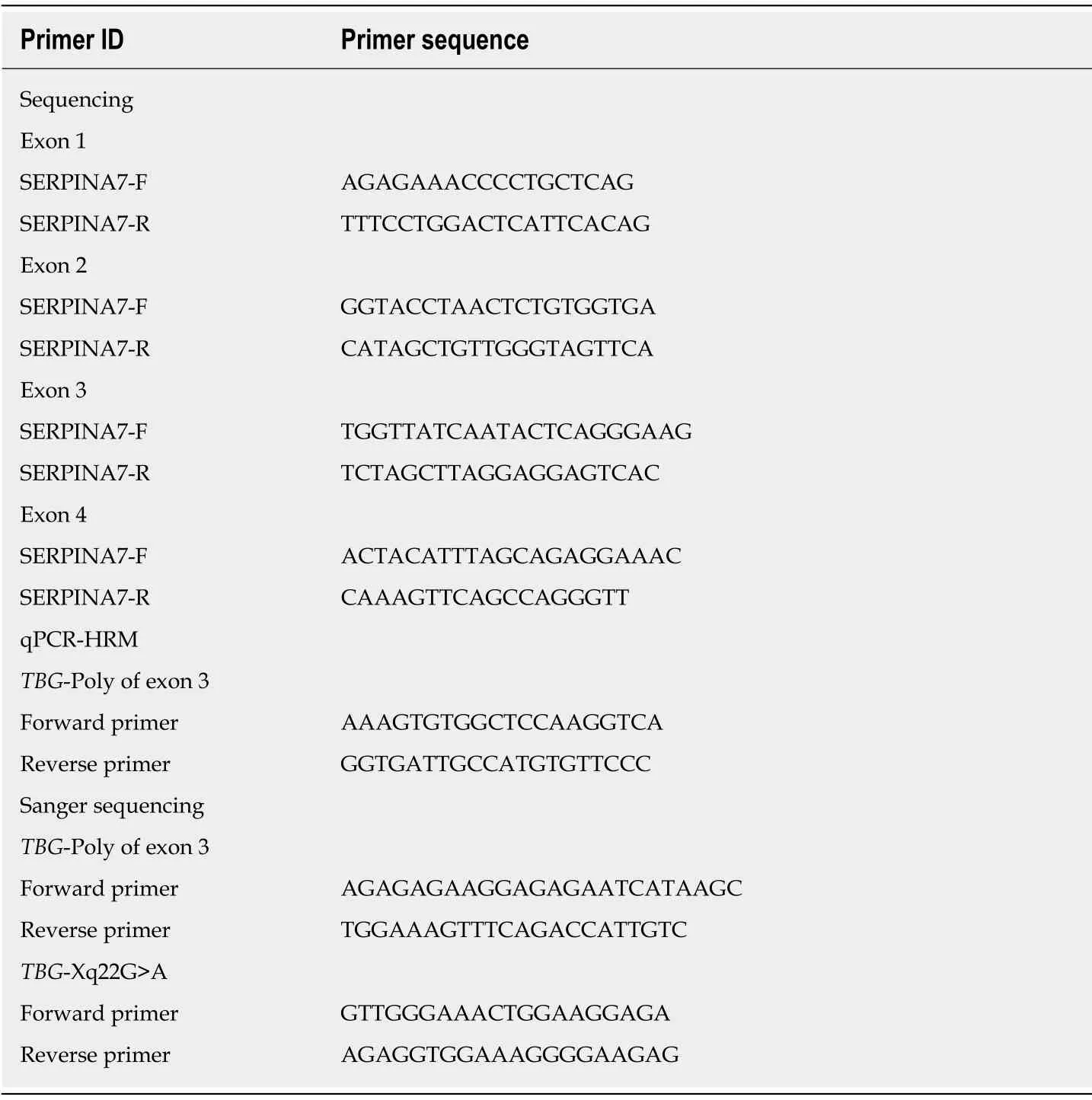
Table 1 Primers used in PCR amplification and sequencing
The frequency of theTBG-Poly allele among the 117 unrelated Han Chinese men was found to be 21.37%.
DISCUSSION
In the present study, we identify a novel mutation, named TBG-CDC, inTBG(p.Phe135Alafs*21) from a Chinese family.This mutation is a 19-nucleotide insertion,located between cDNA positions 381 and 382 in exon 1, and is the first reported large nucleotide fragment insertional mutation of theTBGgene that results in TBG-CD.
This mutation resulted in a truncated protein that contained only the first 134 of the 395 amino acids of the mature TBG-C, and lacked 66% of the carboxyl terminal amino acids.The carboxyl terminus of TBG is important for intracellular transport and protein synthesis[15].We used Mutation Taster software to predict the disease-causing potential of this mutation[16].The prediction suggested that the truncated TBG protein lost two glycosylation sites (at positions 165 and 253), and two thyroxine binding sites(at positions 293 and 398).Glycosylation plays an important role in the processing,folding, and secretion of TBG, and the thyroxine binding sites of TBG are associated with the transport of thyroid hormone in blood[17-19]; we therefore speculate that this truncation of TBG may lead to TBG-CD, and may result in a reduction in TT3and TT4levels.However, the mechanism for the failure to detect immunoreactive TBG in the serum of these patients harboring this mutation remains unknown.We speculate that three reasons may be concerned.The first one may be the impaired synthesis of the truncated TBG proteins.The 19-nucleotide insertion in exon 1 is supposed to affect the transcription or the translation of TBG, leading to the failure of synthesis of mutant TBG.The second one may be the impaired secretion of the truncated TBG proteins.Most of the truncated TBG molecules previously reported were not secreted[20-22].They remained in the rough endoplasmic reticulum and were rapidly degraded within the cells, or had impaired intracellular transport in the blood.And the last reason may be associated with the TBG antibodies used in the present study, which can only bind to the truncated region, but not to the non-truncated region, and hence results in no detection of serum TBG.
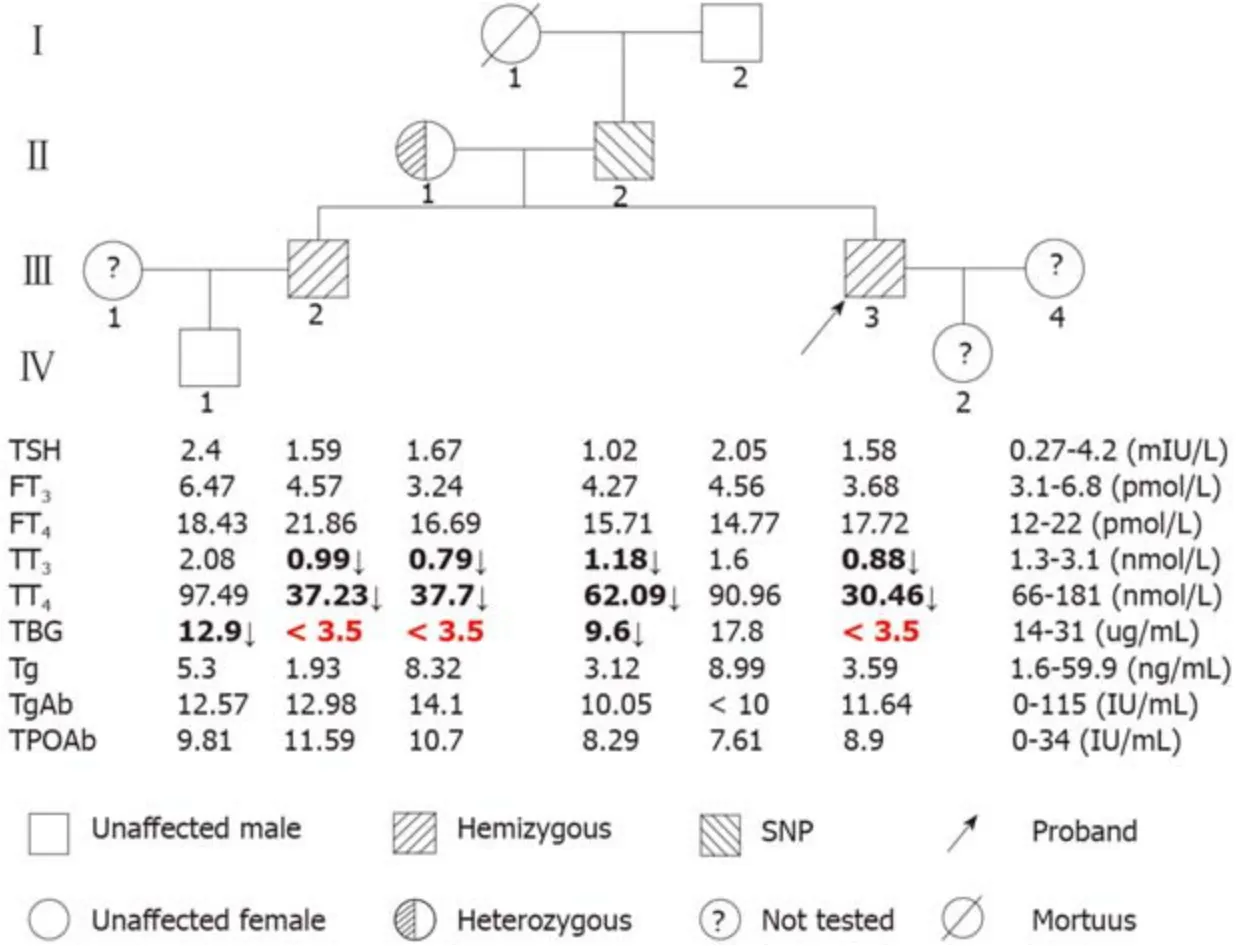
Figure 1 Pedigree showing the genotype and thyroid function test results of the proband’s family.
Since inherited TBG defects follow an X-linked pattern, TBG-CD is fully manifested in hemizygous males but only partially in heterozygous females[1].However, the TBGCD phenotype has been reported in two heterozygous females with selective inactivation of the X-chromosome carrying the normalTBGalleles[23], and in two females with XO Turner’s syndrome[24,25].
In the family we studied, the II-1 also manifested TBG-CD, but the molecular basis for her TBG-CD remains unclear.The proband’s mother has normal stature and fertility, and therefore XO Turner’s syndrome is unlikely, although the condition cannot be ruled out without additional investigation.We speculate that selective inactivation of the X-chromosome containing theTBG-C allele may be associated with her phenotype presentation.
TheTBG-Poly (L283F) variation is a common polymorphism, with high prevalence in different races and regions, including 16% of French Canadian males[26], 50% of Australian Aboriginal males[27], 20% of the Japanese population[28], 31% of the Han Chinese population in Taiwan[29], and 21.37% of Chinese men in northeastern China.Previous studies have reported thatTBG-Poly (L283F) causes no changes in biological properties[27].It is worth noting that the majority of complete TBG defects are associated with nonsense mutations that produce truncated protein, or with missense mutations associated with the presence of the L283F polymorphism (TBG-Poly)[26,29].In the present study, we report that the II-2 presented with the TBG–PD phenotype.Whether theTBG-Poly (L283F) is responsible for lower TBG levels as well as lower levels of TT3and TT4remains unclear.Ferraraet al[12]previously reported a mutation in theTBGgene enhancer region, the Xq22G>A mutation, which is located 20 kb downstream of theTBGgene; this mutation has been associated with the TBG-PD phenotype.However, the Xq22G>A mutation was not detected in the II-2.Since onlyTBGcoding regions and adjacent intron regions were sequenced in the present study,abnormalities in other transcriptional regulatory elements associated with this gene might be involved in the variable phenotypes seen in the Chinese male withTBG-Poly(L283F); these possibilities merit further investigation.
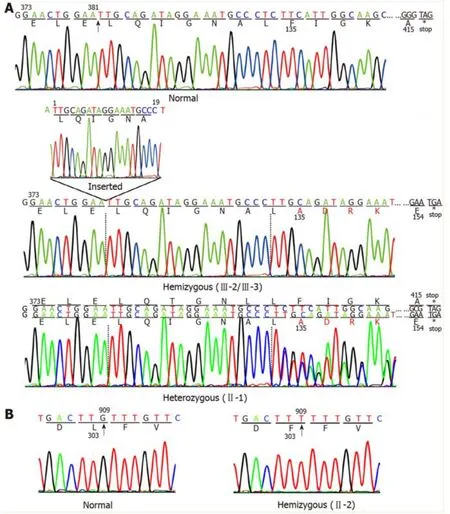
Figure 2 Schematic diagram of the DNA sequence for a portion of exons 1 and 3 of the TBG gene.
TBG deficiency may produce alterations in total thyroid hormone concentration in serum, whereas free THs remain unchanged.So TBG deficiency is often misdiagnosed as hypothyroidism.Clinical awareness is needed to correctly diagnose affected individuals and avoid inappropriate treatment.Genomic testing is a method to identify the mutation carriers and provide appropriate genetic counseling for affected individual[29,30].
CONCLUSION
In conclusion, a novelTBGmutation, p.Phe135Alafs*21, was identified in a Chinese family.This mutation is a 19-nucleotide insertion in exon 1, produces truncated TBG protein, and is termed “TBG-CDC”.The fact that the proband’s father had theTBGPoly (L283F) variant and presented as TBG-PD merits further investigation.The allelic frequency ofTBG-Poly (L283F) was found to be 21.37% in 117 unrelated Chinese males in northeastern China.
ACKNOWLEDGMENTS
We gratefully acknowledge all of the subjects who participated in this study.
 World Journal of Clinical Cases2019年22期
World Journal of Clinical Cases2019年22期
- World Journal of Clinical Cases的其它文章
- Microbial transglutaminase should be considered as an environmental inducer of celiac disease
- Exogenous endophthalmitis caused by Enterococcus casseliflavus:A case report
- Diffuse large B-cell lymphoma arising from follicular lymphoma with warthin’s tumor of the parotid gland - immunophenotypic and genetic features:A case report
- Apatinib for treatment of a pseudomyxoma peritonei patient after surgical treatment and hyperthermic intraperitoneal chemotherapy:A case report
- Peritoneal cancer after bilateral mastectomy, hysterectomy, and bilateral salpingo-oophorectomy with a poor prognosis:A case report and review of the literature
- Diagnosis of gastric duplication cyst by positron emission tomography/computed tomography:A case report