水合鈾酰與尿嘧啶及其異構體配位體系的計算研究
2019-03-13 03:06:42牟永曉曹建平翁約約王朝杰
無機化學學報 2019年3期
關鍵詞:結構
牟永曉 曹建平 翁約約,2 孫 祥,3 衛 濤 王朝杰*,
(1溫州醫科大學藥學院,溫州 325035)
(2溫州市中心醫院藥劑科,溫州 325000)
(3溫州醫科大學附屬第一醫院藥學部,溫州 325000)
0 引 言
隨著軍事和核能工業的快速發展,如何處理核廢料以及如何避免核污染一直是國際社會共同關注的難題。有數據顯示,現階段全世界443座運行的核反應堆已累積了3.6×105噸致命的高放射性核廢料,并且以每年1.2×104噸的速度增長[1]。傳統核燃料主要為錒系元素中的鈾,是一種放射性損傷核素,半衰期長,造成的核污染可持續數十萬年以上,而且其具有的化學毒性常被輕視。鈾的核外層電子組態為5f36d17s2,自然條件下以六價態形成的鈾酰離子(UO22+)最為穩定[2-3]。UO22+可與多種無機或有機配體形成配合物并逐漸滲入地下水和土壤中[4],最終不可避免地與生物體接觸,對人類和環境具有潛在的危害性。Garmash[5]等通過體外研究發現,低濃度的鈾酰離子會造成DNA和蛋白質結構的氧化損傷,其毒性主要與鈾酰離子物理化學性質有關。具有高靈敏度、高親和力的鈾特異性脫氧核酶,已廣泛應用于環境中鈾酰離子的生物監測[6]。因此研究鈾酰離子與多種生物有機小分子的結合行為有助于我們更深入地了解鈾對生物體的影響。除了實驗研究外,理論計算也是研究鈾酰離子與生物小分子相互作用的重要途徑,可為實驗研究鈾酰離子在生物機體內的吸收、轉運、沉積的分子機制提供理論支持。
目前國內外對鈾酰離子和有機小分子的理論研究已有大量報道。如Zakharieva等[7]應用相對論密度泛函方法研究溶液中鈾酰離子與含氮有機小分子配位形成[UO2(H2O)4(LN)]2+(LN=NH2CH3、N(CH3)3、NC5H5)的相互作用性質,并與五水合鈾酰離子作比較,結果顯示甲胺配合物最穩定,吡啶配合物的能量與五水合鈾酰離子相差不大,而三甲胺配合物的能量比五水合鈾酰離子高約40 kJ·mol-1。Wu等[8]報道了鈾酰離子與絲氨酸和磷酸化絲氨酸在溶液中的配位結構和能量學性質,研究發現體系更傾向于形成五配位鈾酰配合物,且當鈾酰離子與配體以1∶3配位時配合物的結合能最高。Ren等[9]也通過密度泛函理論方法,研究了在水溶液中水合鈾酰離子與氧代二乙酸、亞氨基二乙酸、硫代二乙酸形成飽和配位時的配合物結構特性和電子結構。Vukovic[10]等對水合UO22+與胺肟陰離子配合物進行了理論計算,其中胺肟陰離子中氮和氧在鈾酰的赤道面上發生雙齒配位,并將計算結果與實驗測定UO22+與胺肟及其衍生物陰離子配合物的結果進行了比較。與大多數研究UO22+離子與氨基酸或多肽分子配位特征的理論研究相比,UO22+離子與核酸堿基分子或DNA片段相互作用的體系仍然較少[11-12]。
核酸的生物學行為及生物活性都是以構成其基本單元的堿基分子的性質為基礎,而尿嘧啶(Uracil,Ur)是十分重要的核酸堿基分子,其6種互變異構體的結構和光譜性質已有大量研究報道[13-18]。鈾酰離子介導的光裂解可用于RNA結構的構象分析,如Wittberger等[19]研究發現RNA二級結構的單鏈區域是鈾酰的裂解位點。在tRNA中,鈾酰裂解位點是在環結構內部以及中心鉸鏈區。
本文應用密度泛函理論方法在相對論有效勢基組水平對水合鈾酰離子與尿嘧啶及其異構體形成的一系列配離子的結構與性質進行了計算研究,為進一步研究(脫氧)核苷酸或(脫氧)核糖核酸片段與錒系酰離子的配位行為和機制提供基礎。
1 計算方法
迄今對于含錒系元素體系的計算報道甚多[20-23],以全電子(all-electon,AE)的相對論計算最為認可,但與可用的實驗數據如振動光譜和結構參數比較,也并未體現出絕對優勢[24]而且效率受限,其他模型勢(model potential,MP)或贗勢(pseudopotential,PP)的有效核勢(effective core potentials,ECPs)及其基組得到更多的應用[25],但其計算結果常常與計算體系、計算方法尤其是密度泛函方法的選取有相關[26-29],本工作運用大核贗勢和最廣泛使用的雜化泛函來探索。
選用雜化密度泛函B3LYP方法[30],此泛函是研究鈾酰離子配合物體系常用的計算方法之一[31-35],且表明對錒系酰離子的基態結構描述較為合理。計算中對鈾原子采用Stuttgart RLC ECP[36]贗勢基組,是包含了相對論效應的有效勢和價層DZ基組,其中有效勢對鈾內層電子 1s~5s、2p~5p、3d~5d、4f共78個電子做凍芯處理,外層的14個電子(5f36s26p66d17s2)作為價層電子,其他原子采用6-311++G(d,p)基組。 對[UO2(Uracil)j(H2O)k]2+(Uijk,i=1~6,j=1~5,k=5-j,i為6種尿嘧啶異構體代號,j表示尿嘧啶配位數,k表示水分子配位數)體系的幾何結構參數、振動光譜和能量學等性質進行計算,振動光譜分析均沒有虛頻,可確認其為穩定結構。
分子中的原子理論(Quantum theory of atoms in molecules,QTAIM)用于分析體系中配位作用和電荷轉移變化。根據Bridgeman等[37]研究結果,在計算Mayer鍵級和QTAIM分析時對其他原子宜采用6-311G(d,p)基組。采用極化連續介質模型(polarized continuum model,PCM)[38]模擬水溶液對配離子的溶劑化效應。所有計算工作使用Gaussian09[39]程序包結合Multiwfn3.5[40]程序完成。
2 結果與討論
2.1 水合鈾酰尿嘧啶配離子的結構性質
首先我們對6種尿嘧啶異構體作了B3LYP/6-311++G(d,p)水平的優化計算,其幾何結構參數和相對能量數據列于支持信息圖S1和表S1,按氣相相對吉布斯自由能由低到高的順序依次編號為Ur1~Ur6,6種異構體均含有羰基和(或)羥基基團且只連接在C2和C4位上,其中Ur1屬于雙酮式,Ur4為雙羥基結構,其余4種異構體之間的差別在于羥基和羰基的連接位置不同(如Ur2與Ur6、Ur3與Ur5)。最穩定的尿嘧啶(Ur1)比羰基-烯醇式異構體(Ur2)能量低48.4 kJ·mol-1,6種尿嘧啶異構體穩定性順序與文獻報道的計算結果一致[15-16]。
據相關鈾酰離子配位體系的研究報道,當配體在鈾酰軸的赤道面上與鈾原子形成五配位鍵時可達飽和配位,且該飽和配合物在氣相和水溶液中均比對應的不飽和配位結構穩定[41-44]。計算尿嘧啶Ur1與水合鈾酰五配位的結構(U1jk),得到14種穩定結構(支持信息圖S2)。從圖S2可知,Ur1與鈾酰離子的相互作用有2種類型,即環上的羰基氧(C2=O7和C4=O8)分別與鈾原子配位形成U-O配位鍵。表S2列出了14種配合物的相對吉布斯自由能。結合圖S2和表S2,當Ur1以C4=O8參與配位時,配離子能量最低,與對應的以C2=O7鍵參與配位的結構相比,兩者能差在34.7~88.9 kJ·mol-1范圍內。 而配位鍵類型相同,配體間空間位置不同的結構能量十分接近,后文中我們只對比分析最穩定的配合物結構。
五水合鈾酰離子及U1jk體系在氣相和水相中計算得到的U=O鍵長值接近于實驗[45]測定值(0.170 2 nm)。文獻中運用相同方法下計算其它體系得到的U-OH2鍵長值在0.252 0~0.265 6 nm范圍[46-47],與我們結果一致。Ur1羰基氧與鈾原子的配位距離為0.230 1~0.239 9 nm,與實驗[48]測得的其它酮類結構(如 N,N,N′,N′-四正丁基丙二酰胺)中的羰基氧與UO22+的配位距離(0.236 0 nm)相近。
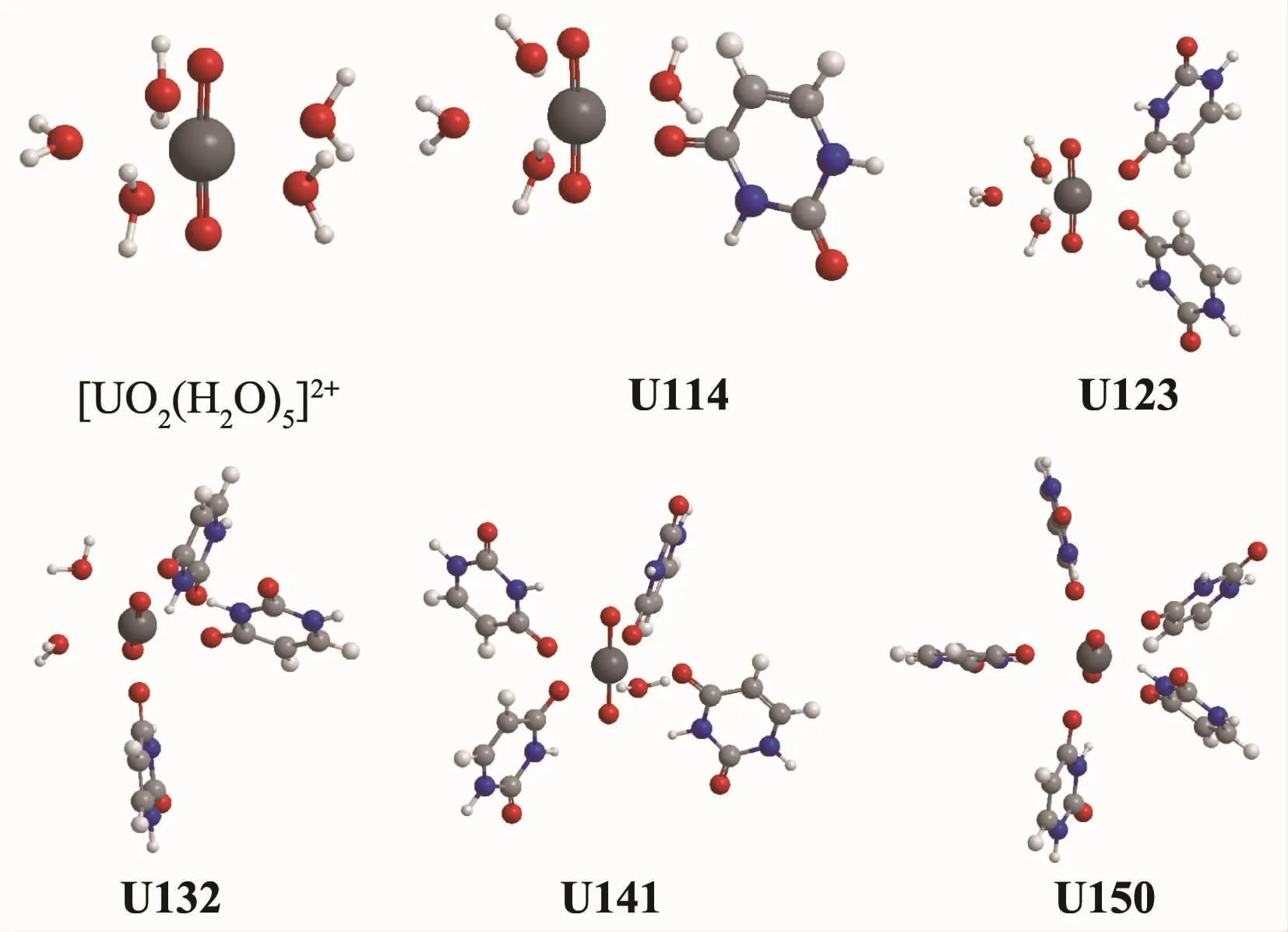
圖1 五水合鈾酰離子和水合鈾酰尿嘧啶配離子[UO2(Uracil)jH2O)5-j]2+(j=1~5)在氣相中的平衡結構Fig.1 Equilibrium structures of uranyl pentahydrate and hydrated uracil coordination uranyl ions[UO2(Uracil)j(H2O)5-j]2+in gaseous phase
隨著Ur1配位數目的連續增加,配離子中U=O鍵、U-OH2鍵和U-Ouracil鍵均有不同程度的伸長,說明配體與中心鈾原子間的作用整體上呈變弱的趨勢。例如U=O鍵長從五水合鈾酰離子的0.172 4 nm增長至U150配離子的0.176 0 nm,對應的Mayer鍵級則從2.35逐漸降低至2.14。另外U-Ouracil鍵長從 0.230 1 nm(U114)增長至 0.241 9 nm(U150),對應的鍵級則從0.53降至0.39,U-OH2鍵長從五水合鈾酰離子的0.252 3 nm增長至U141體系中的0.258 7 nm,鍵級則從0.31減弱為0.26。U-Ouracil鍵長均比U-OH2鍵短,反映出尿嘧啶與鈾酰離子的作用強度大于水分子與鈾酰離子的作用強度。
考慮溶劑化效應后,在水溶液中U=O鍵和UOuracil鍵長伸長,而U-OH2鍵長縮短,在文獻中也同樣發現溶劑化后U=O鍵和U-Oligand鍵減弱,但U與水分子的作用增強[42,49],可能由于水環境會使得水的電子分布被極化,導致水中氧的孤對電子效應增加,加強了與鈾原子的靜電作用。在Ur1配位數為1~2的結構中,U-Ouracil鍵增長明顯,平均增長了0.034 5 nm。而在U141和U150結構中,水溶液與氣相中的U-Ouracil鍵長接近。表明在高配位數下,溶劑對鈾酰尿嘧啶的配位影響逐漸減小。
一分子的尿嘧啶異構體與水合鈾酰離子作用形成的飽和Ui14結構如圖2所示。尿嘧啶異構體與中心鈾原子存在3種配位類型:(1)羰基O與U的單配位, 如結構 U114、U214、U314、U514, 其中Ur1、Ur2、Ur3的環平面與鈾酰軸平行,Ur5的環平面與鈾酰軸垂直;(2)羥基O的單配位,如結構U414;(3)羰基O和環上N的雙齒配位,如結構U613,由于飽和配位數是5,水分子配體數為3。Ui14體系的相對自由能數據列于支持信息表S3。從表S3可知,在氣相中的相對能由低到高順序為U514<U314<U114<U214<U414,因 U613 水分子配體數目不同,故不將U613結構納入相對能量的比較。在結構U514與U314中,Ur5、Ur3環上的N均與鄰位的水分子形成了N…H-O氫鍵,彼此氣相吉布斯自由能量相差僅為1.5 kJ·mol-1,而U214的相對能差高達 37.3 kJ·mol-1。
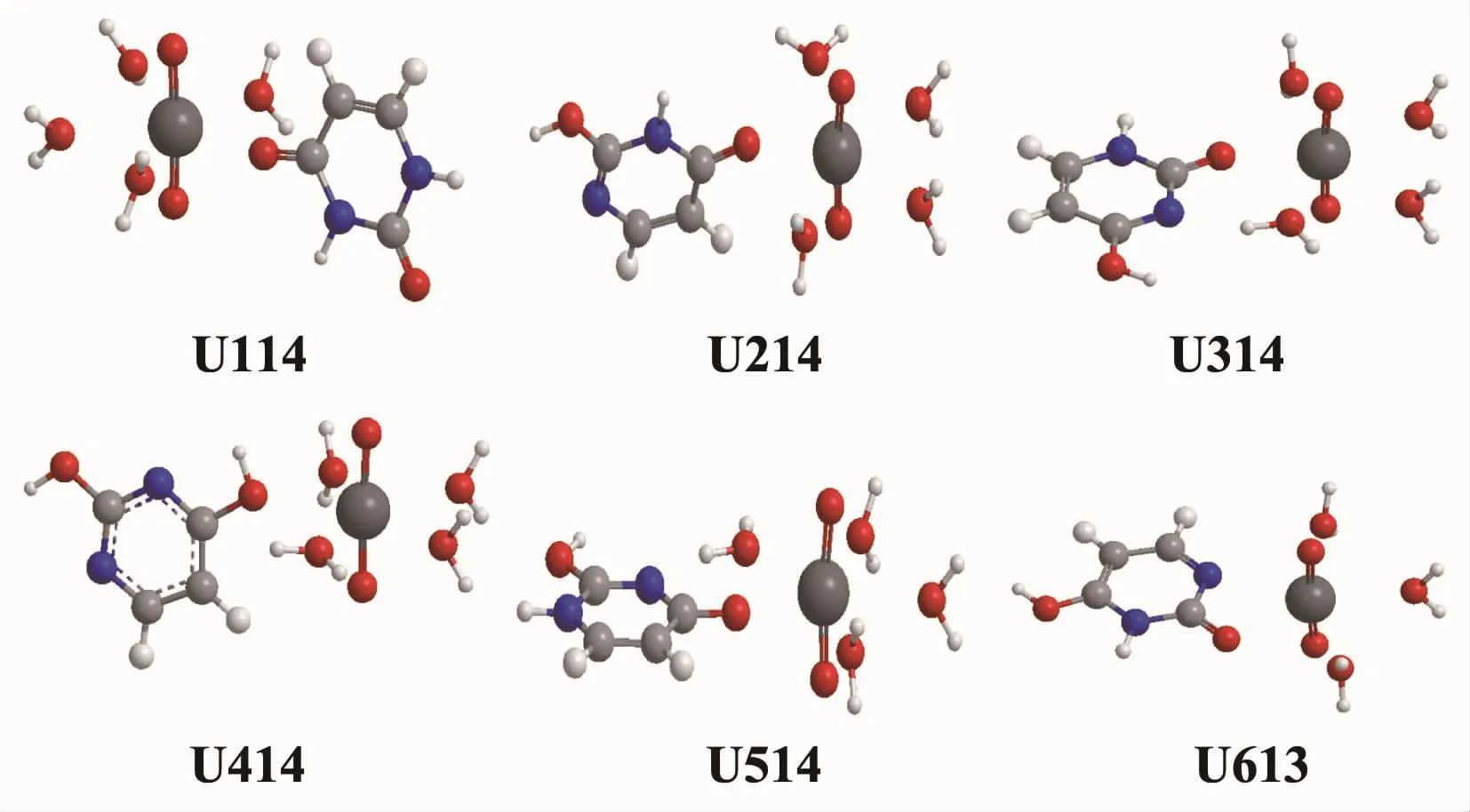
圖2 水合鈾酰離子與6種尿嘧啶異構體在氣相中形成1∶1型飽和配離子的平衡結構Fig.2 Equilibrium saturated structures of 1∶1 type complexes of hydrated uranyl ion with six kinds of uracil tautomers in gaseous phase
Ui14體系的幾何結構參數列于表1中,表中數據表明U=O鍵的鍵長在各體系中較為接近。U-Ouracil鍵的鍵長遞增順序為 U214(0.229 9 nm)<U114(0.230 1 nm)<U514(0.230 5 nm)<U314(0.234 1 nm)<U613(0.243 5 nm)<U414(0.250 4 nm),對應的鍵級大小順序則相反,而U-OH2鍵距按U613(0.251 6 nm)<U414(0.252 5 nm)<U314(0.253 9 nm)<U514(0.254 5 nm)<U114(0.255 6 nm)<U214(0.255 6 nm)的順序遞增。可見U-Ouracil鍵長與U-OH2鍵長整體上呈相反變化,表明尿嘧啶異構體與水分子配體存在明顯配位競爭作用。Ui14結構U-OH2的平均鍵級為0.29,與文獻[39]報道的四水合過氧鈾酰[UO2(O2)(H2O)4]2+中U-OH2的鍵級(0.30)相接近。值得指出的是,U414結構U-Ouracil的鍵級為0.28,顯著低于其它5種結構,且它與U514的氣相吉布斯自由能相差高達182.9 kJ·mol-1,同時在水溶液環境的優化計算中,U414結構無法正常收斂,可見在水溶液中不能穩定存在。通過對相對能量和鍵級的分析,表明異構體Ur4與鈾酰離子的作用強度較弱,由于Ur4是以羥基氧配位。考慮溶劑化效應后,各結構U-Ouracil鍵和U-OH2鍵的鍵長遞增順序較氣相下發生改變。

表1 在氣相和水相中水合鈾酰尿嘧啶及其異構體配離子的結構參數和Mayer鍵級Table 1 Equilibrium structure parameters(nm)and Mayer bond orders of hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
2.2 水合鈾酰尿嘧啶及其異構體配離子的振動光譜
U1jk結構中主要鍵的振動頻率計算結果列于表2。由于B3LYP方法的基頻校正因子都接近于1[50],因此不考慮基頻校正因子。從表2中可知,五水合鈾酰離子中O=U=O鍵的振動頻率為對稱(νss)961 cm-1和反對稱(νas)1 039 cm-1,高于文獻中[49]通過B3LYP/6-31+G**(RECP)水平計算得到的O=U=O鍵對稱(937 cm-1)和反對稱(1 026 cm-1)伸縮振動頻率。水溶液中O=U=O鍵的伸縮振動頻率與實驗測定值[51](νss870 cm-1,νas965 cm-1)相差在 67 cm-1范圍內。
在水溶液中,當Ur1分子逐步取代水分子后,O=U=O鍵對稱和反對稱伸縮振動頻率逐漸降低,表明配位體系穩定性增加[52],辜家芳等[47]的報道也證實了同樣的結論。因此,結合前文幾何結構分析,說明尿嘧啶分子比水分子更易于與鈾酰離子結合。與自由狀態下Ur1環上氨基鍵(N-H)的伸縮振動頻率3 616 cm-1相比(表S4),配位結構中的Ur1環NH鍵發生紅移,且紅移范圍在45~50 cm-1間,說明羰基氧的配位削弱了鄰位的N-H鍵。
計算的鈾酰尿嘧啶異構體配離子(Ui14)相關鍵的振動頻率數據也列于表2中,配位的O=U=O伸縮振動頻率較為接近(除了U414結構)。6種結構的O=U=O鍵對稱伸縮振動頻率按照U214(935 cm-1)<U514(938 cm-1)<U114(939 cm-1)<U314(941 cm-1)<U613(946 cm-1)<U414(949 cm-1)的順序遞增,對應結構的U-Ouracil鍵長整體也呈變長的趨勢。U414結構O=U=O鍵的伸縮振動頻率與[UO2(H2O)5]2+結構伸縮振動頻率相接近,說明尿嘧啶環的羥基氧與鈾酰離子的相互作用較弱,與前文幾何結構分析結果一致。考慮溶劑效應后,發現水相結構中O=U=O鍵的伸縮振動頻率紅移,而νN-H藍移。
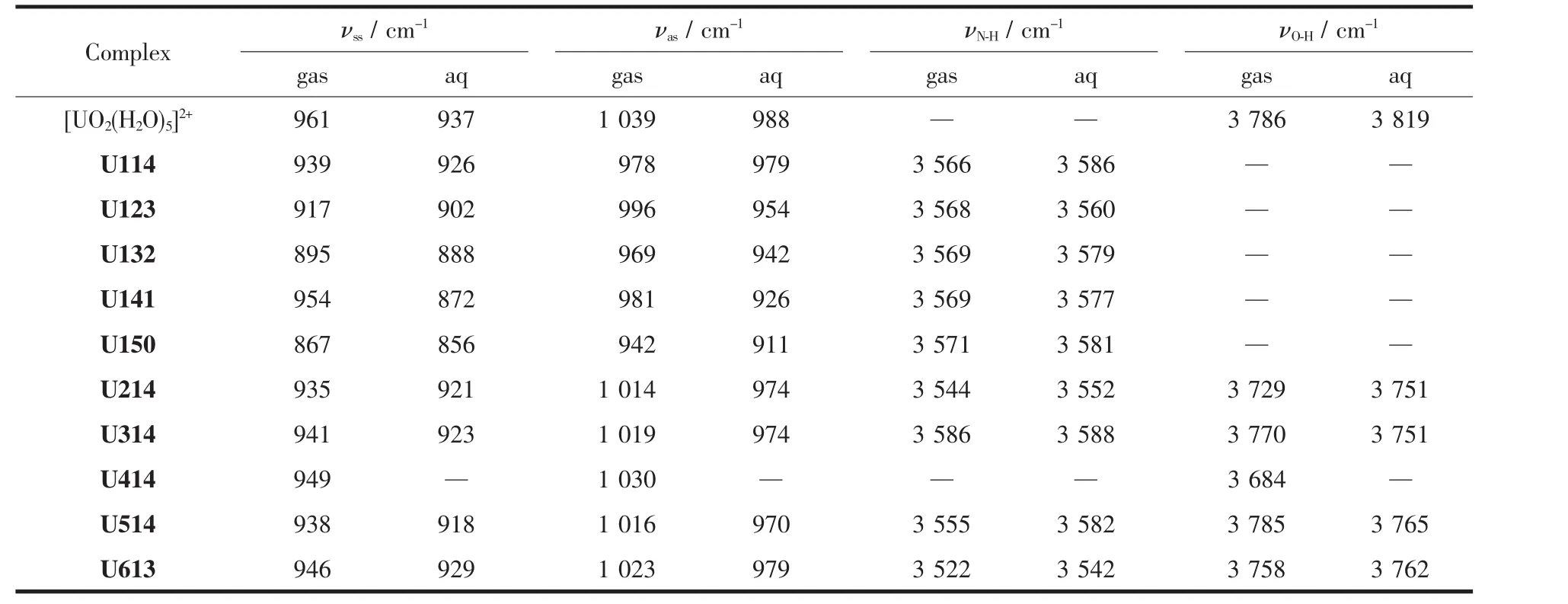
表2 在氣相和水溶液中水合鈾酰尿嘧啶及其異構體配離子中UO22+的對稱(νss)和反對稱(νas)伸縮振動頻率、尿嘧啶及其異構體環上N-H鍵和O-H鍵的伸縮振動頻率Table 2 Vibrational frequencies(νssand νas)of U=O bond and vibrational frequencies of N-H and O-H bonds of uracil and its tautomers in hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
2.3 水合鈾酰尿嘧啶及其異構體配離子的能量學分析
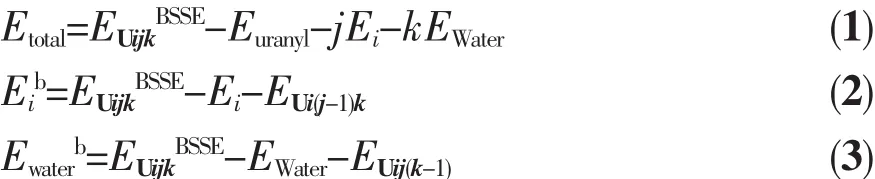
為了分析配離子穩定性及配體與鈾酰離子的結合強度,計算了Uijk體系的總結合能及尿嘧啶和水配體分別與鈾酰離子作用的單結合能,單結合能計算類似于“第一電離能”的計算方法,計算公式如下:將配位體系分為鈾酰離子、尿嘧啶、水分子3個片段,其中EUi(j-1)k、EUij(k-1)分別表示失去一分子尿嘧啶和一分子水的配合物的能量,而Eib、Ewaterb表示尿嘧啶和水分子的單結合能,即在配離子中單個配體的結合能量。計算以上相互作用能時均考慮了基組重疊誤差(basis set superposition error,BSSE)校正。 計算得到的各項結合能數據列于表3中。從表3可知,氣相中[UO2(H2O)5]2+的結合能為-1038.0kJ·mol-1,Spencer等[53]在 BLYP/DZP(ECP)計算水平,不考慮BSSE校正得到[UO2(H2O)5]2+的結合能為-1 073.6 kJ·mol-1,與本文計算的結合能相一致。
在U1jk系列結構中,隨著尿嘧啶配體數目的增加,總結合能逐漸增大,配體的單結合能在逐漸減小。U-Ouracil鍵的結合能比U-OH2鍵的結合能大,體現尿嘧啶與鈾酰離子的作用比水分子與鈾酰離子的作用強,但是與文獻[47]中計算得到的鈾酰酸根配 合 物[UO2(CO3)2]2-(-688 kJ·mol-1)和[UO2(NO3)3]-(-367.7 kJ·mol-1)的單結合能相比較,發現尿嘧啶的單結合能較和的小,這表明帶負電荷的酸根陰離子比中性尿嘧啶分子更易與鈾酰離子發生相互作用。在水溶液中,水分子的單結合能低于氣相下的結合能,說明U-OH2配位鍵的鍵能降低,體現出溶劑效應。
為了定量描述結合能與Ur1配體數量(0~5)間的關系,我們使用Origin軟件對U1jk體系的結和能進行擬合,擬合曲線如圖3所示。由于在水溶液中配體單結合能的變化趨勢不明顯,故只對氣相下的單結合能進行擬合分析。在氣相下,得到尿嘧啶配位數(j)與總結和能(ETotalgas)的擬合結果為:
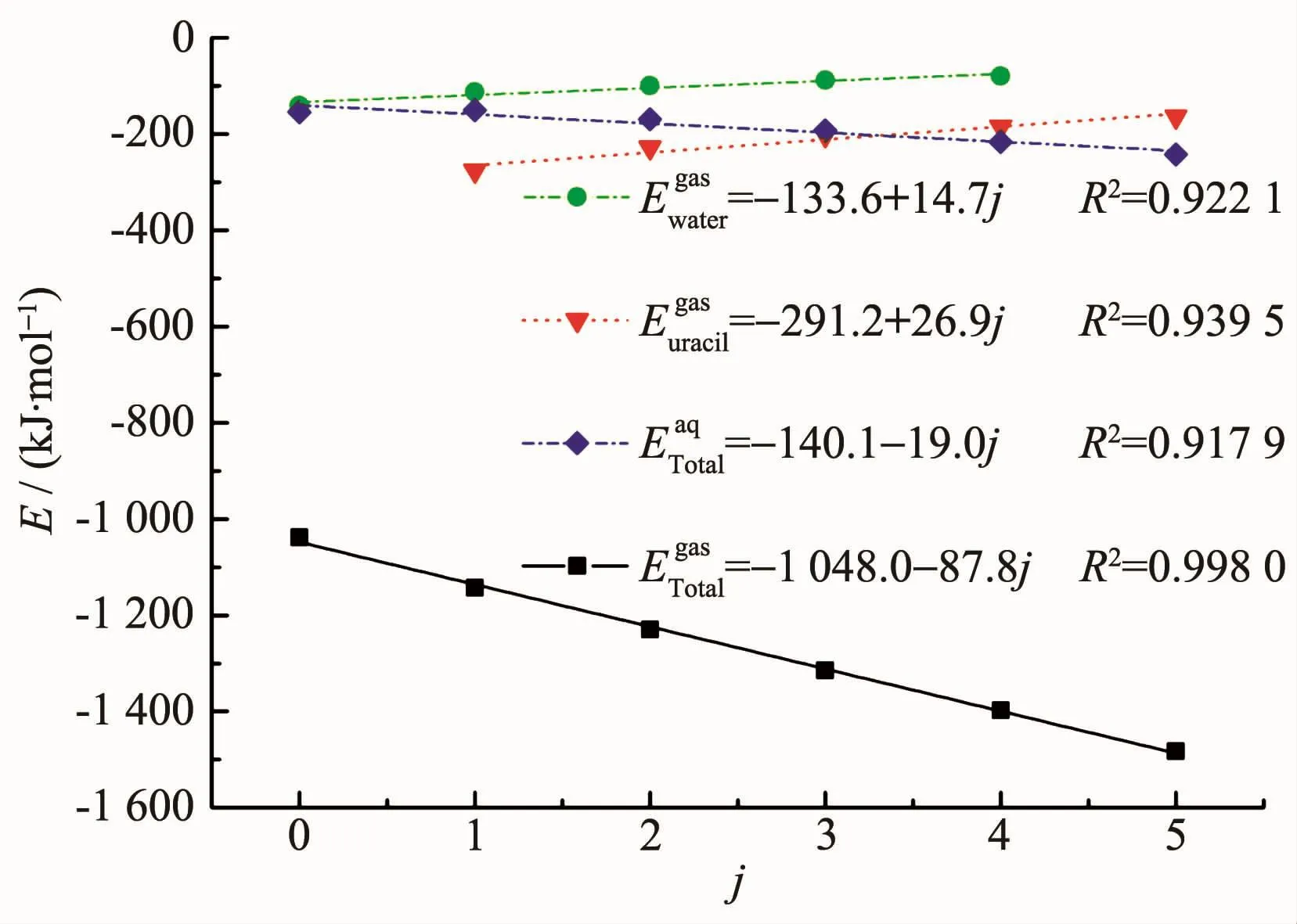
圖3 U1jk體系在氣相和水溶液中結合能與Ur1配位數j的關系Fig.3 Relationships between binding energies and the number j for Ur1 ligand of U1jk systems in gaseous and aqueous phases
在水溶液中,得到j與ETotalaq的擬合結果為:

可見氣相下總結和能的擬合度R2值更接近1,表明氣相下線性擬合的效果較好。水溶液中的總結和能增加量較小,為19.0 kJ·mol-1,這可能是由于Ur1分子極性較大,嘧啶環上的氨基H和羰基O均可與水分子形成氫鍵,故考慮溶劑化效應對結合能的變化影響較小。
更負的結合能表明配體與鈾酰有更強的相互作用,體系亦更穩定。比較Ui14體系的總結合能在-1 014.1~-1 238.5 kJ·mol-1的范圍,見表 3。 Ui14體系中結合最緊密的(U514)不是由能量最低的Ur1異構體生成,但是Ur5本身存在的概率較低。U514結構的總結合能在氣相和水溶液中均最負,這與Ur5環上的N與相鄰的水分子形成了配體間氫鍵相關,協同增強。U613結構中尿嘧啶的單結合能(-432.5 kJ·mol-1)最大,體現配體與鈾酰離子配位的成鍵數目越多,兩者的相互作用越強。U414結構中尿嘧啶的單結合能(-143.6 kJ·mol-1)與水分子的單結合能(-133.2 kJ·mol-1)略接近。由于溶劑效應的影響,水溶液中配體的單結合能顯著減小。
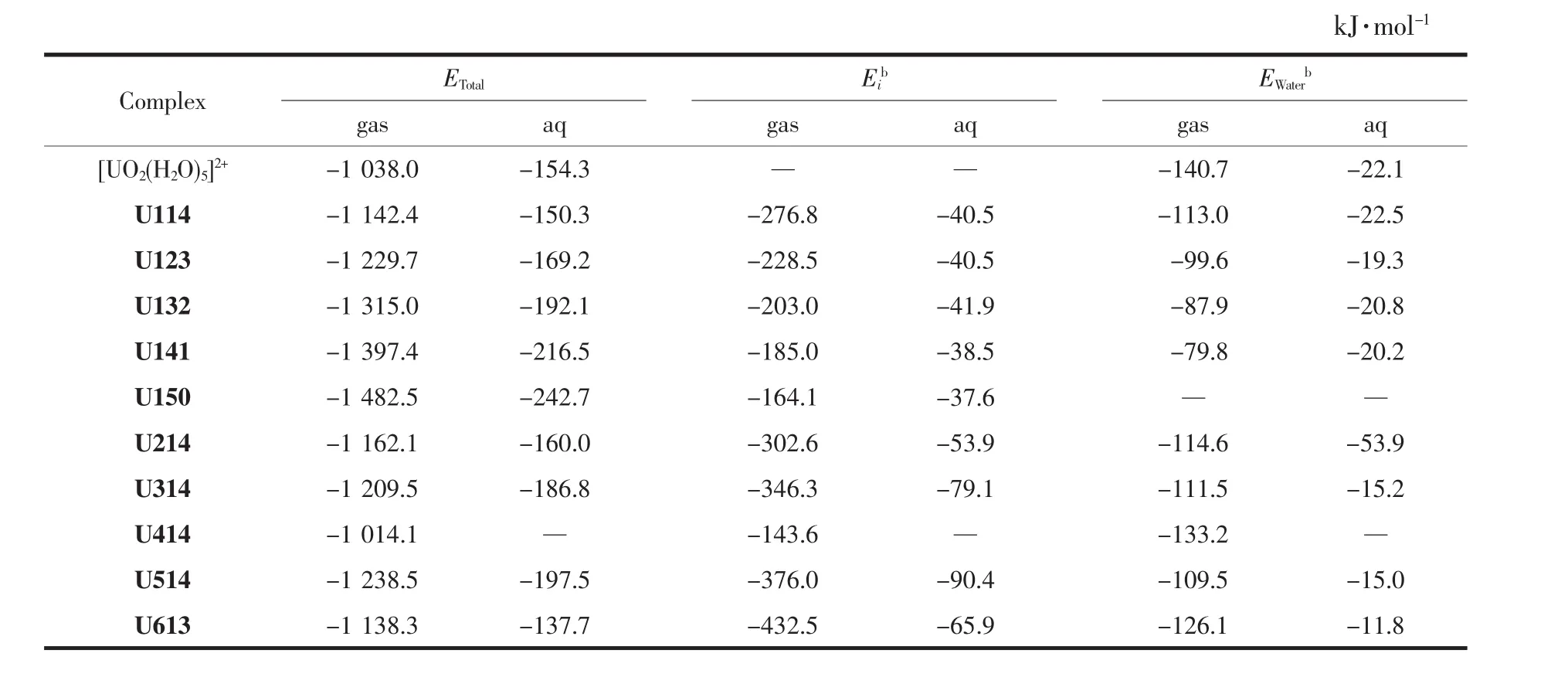
表3 在氣相和水溶液中水合鈾酰尿嘧啶及其異構體配離子的結合能EBSSETable 3 Binding energy with the BSSE correction for hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
2.4 配離子的拓撲分析
為了進一步研究體系中配位鍵的形成和強度,我們使用Multiwfn程序進行QTAIM拓撲分析。根據 Bader[54]理論,當 BCP處的 ρ(r)大于 0.2 a.u.且▽2ρ(r)為負值時,說明該鍵具有共價鍵性質,若BCP處的 ρ(r)小于 0.2 a.u.且▽2ρ(r)為正值時,則該鍵具有離子鍵性質。表4列出了氣相中配位鍵在(3,-1)臨界點處的電子電荷密度和電子密度拉普拉斯值。從表 4中 U1jk體系的 U-Ouracil鍵在 BCP處的 ρ(r)和▽2ρ(r)分別在 0.058~0.072 a.u.和 0.236~0.326 a.u.范圍內,U-OH2鍵在 BCP 處的 ρ(r)和▽2ρ(r)分別在 0.039~0.043 a.u.和 0.156~0.174 a.u.范圍內,說明 U-Ouracil鍵和U-OH2鍵具有離子鍵的特征。文獻[8]報道了水合鈾酰離子與絲氨酸分子相互作用形成的U-Ocarboxyl配位鍵在 BCP 處的 ρ(r)約為 0.046,且▽2ρ(r)均為正值,同樣具有離子鍵特征,與本文U-Ouracil鍵的QTAIM分析結果一致。U-Ouracil鍵和U-OH2鍵的ρ(r)和▽2ρ(r)數值整體上隨著Ur1配體數目的增加而逐漸減小,表明其離子鍵特征在逐漸減弱,在Ur1配位數由 3 增加到 4 時,U-OH2鍵的 ρ(r)和▽2ρ(r)值反而略有增大,與上文在氣相中對應的鍵長變化趨勢一致。
Ui14體系中U-Ouracil配位鍵在BCP處的ρ(r)按照 U214(0.075 a.u.)>U514(0.073 a.u.)>U114(0.072 a.u.)>U314(0.068 a.u.)>U613(0.063 a.u.)>U414(0.050 a.u.)的順序遞減,U214、U514、U114 的 ρ(r)值相近,而 U-Ouracil鍵的 ρ(r)和▽2ρ(r)的大小順序與前文的尿嘧啶異構體單結合能強弱順序不同。6種配離子中U-OH2鍵的 ρ(r)范圍在 0.043~0.047 a.u. 之間。Lipkowski等[55]提出了氫鍵的鍵臨界點 ρ(r)和▽2ρ(r)的范圍標準,分別在 0.002~0.040 a.u.和 0.020~0.150 a.u.之間。U514和U314結構中,尿嘧啶環上N與鄰位水分子形成的N…H-O鍵在BCP處的ρ(r)和▽2ρ(r)在上述氫鍵范圍內,可確認其形成了氫鍵。
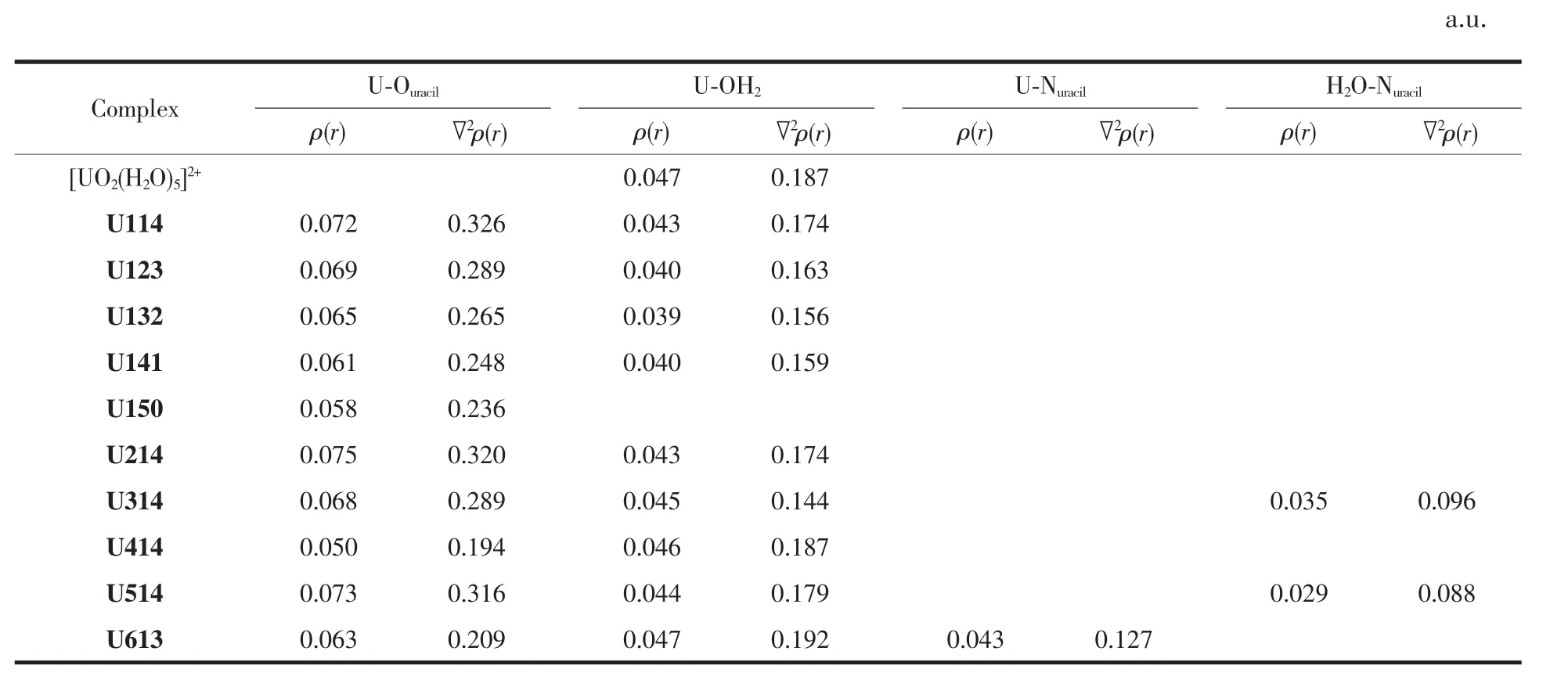
表4 氣相水合鈾酰尿嘧啶及其異構體配離子配位鍵臨界點處的電子密度(ρ(r))和拉普拉斯值(▽2ρ(r))Table 4 Electron density(ρ(r))and Laplacian of electron density(▽2ρ(r))at U-O(N)and H2O-N bondcritical point of hydrated uracil and its tautomers coordination uranyl ions in gaseous phase
2.5 電荷轉移分析
配體間因相互作用而重新平衡后,不僅幾何結構會發生改變,其原子上的電荷也會發生重新分布。為了進一步研究目標體系的電子結構性質,我們對相互作用的3個配體片段(鈾酰離子、尿嘧啶及其異構體、水分子)進行了自然布居分析(natural population analysis,NPA),以此來考察在形成配離子前后各配體片段的電荷分布變化情況。
對于U1jk體系,表5列出了在氣相和水溶液中形成配離子后3個配體片段所帶的NPA電荷。與自由狀態下的片段電荷相比,可以看出形成配離子前后各配體所帶平均電荷變化明顯,其中尿嘧啶和水分子片段所帶平均電荷數目變大,如在氣相中分別增加了0.307~0.383和0.185~0.193。而鈾酰離子所帶電荷數目從自由狀態下的2.000降低至0.465~0.845范圍內。從鈾酰離子片段電荷的降低(得電子)和配體片段的電荷升高(失電子)可說明在配合過程中是由配體片段向鈾酰離子發生了電子轉移。同時,尿嘧啶向中心受體轉移的電子數大于單個水分子向中心受體轉移的電子數。配體向中心受體轉移的電子數越多,形成的配位鍵越強。在U1jk體系中,隨著Ur1配體數量的增加,鈾酰離子所帶電荷數從0.845逐漸減小至0.465,即得到的電子數逐漸增加(1.155至1.535),而尿嘧啶片段的平均電子轉移數從0.383減少至0.307。考慮了溶劑效應的影響后,Ur1片段的電荷變化量略有下降,而水分子所帶的電荷數略升高,此與前文在水環境下UOuracil鍵和U-OH2鍵的鍵長互有增減的結果相一致。表明溶劑效應會影響配離子中配體間的電荷重新分布。
對于Ui14體系,各配體片段所帶的電荷列于表5中。鈾酰離子所帶電荷數按照U214(0.831)<U514(0.837)<U114(0.845)<U314(0.853)<U613(0.857)<U414(0.908)的順序遞增,表明Ur2異構體和水分子向中心鈾酰的電子轉移數最多。而U613結構中尿嘧啶異構體片段所帶電荷數最高 (0.507),Ur6異構體失電子數最多,進一步表明該異構體與鈾酰形成的雙配位鍵會增加電子向中心片段的轉移量。

表5 在氣相和水溶液中水合鈾酰尿嘧啶及其異構體配離子中各配體的自然布居(NPA)電荷數Table 5 Natural population analysis(NPA)for charges of each ligand in hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
3 結 論
采用B3LYP雜化密度泛函方法在相對論有效勢基組和6-311++G(d,p)水平下對[UO2(Uracil)j(H2O)k]2+(j+k=5)配位體系進行研究。對幾何結構、振動光譜、結合能、電子密度拓撲參數、原子電荷轉移等進行了闡述,得到以下幾點結論:(1)配離子結構表明配位鍵及U=O鍵的鍵長隨Ur1配體數目的增加而伸長,且U-Ouracil鍵長較U-OH2鍵短。Ui14體系中U214的U-Ouracil鍵長最短。鍵長大小與Mayer鍵級成負相關關系;(2)振動光譜分析表明,在水溶液中O=U=O鍵的伸縮振動頻率隨Ur1配體數量的增加而減小,較氣相下發生紅移,而νN-H發生藍移;(3)結合能計算表明配離子的總結合能隨Ur1配體數目的增加而增加,但配體單結合能逐漸減小。Ui14結構中U514的結合能最大,并與結合能最低的U414結構相差232.6 kJ·mol-1,可見Ui14體系中結合最緊密的結構并不是由能量最低的Ur1異構體生成;(4)QTAIM拓撲分析表明配位鍵具有離子鍵性質,且臨界點的 ρ(r)和▽2ρ(r)整體上隨著 Ur1配體數目的增加而減小,鍵能降低,與配體的鍵長變化趨勢一致;(5)原子電荷分析明確了在配合過程中是由配體片段向鈾酰發生了電子轉移,且尿嘧啶及其異構體所帶電荷量大于單個水分子的電荷量,同時尿嘧啶的所帶電荷量與配體數量大致成負相關關系。由于大核贗勢在軌道作用分析方面存在不足,在后續錒系酰離子與其它堿基、堿基對、(脫氧)核苷酸或(脫氧)核糖核酸片段作用體系的探索中將對大小核贗勢進行對比研究。
Supporting information is available at http://www.wjhxxb.cn
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
