Gerstmann-Str?ussler-Scheinker disease: A case report
2019-02-22 07:33:50Ming-MingZhao,Liang-ShuFeng,ShuaiHou等
World Journal of Clinical Cases 2019年3期
Abstract BACKGROUND Gerstmann-Str?ussler-Scheinker (GSS) disease is an inherited prion disease that is clinically characterized by the early onset of progressive cerebellar ataxia. The incidence of GSS is extremely low and it is particularly rare in China. Therefore,clinicians may easily confuse this disease with other diseases that also cause ataxia, resulting in its under-diagnosis or misdiagnosis.CASE SUMMARY Here, we report the first case of genetically diagnosed GSS disease in Northeast China. The patient exhibited typical ataxia and dysarthria 2.5 years after symptom onset. However, magnetic resonance imaging of the brain and spinal cord revealed a normal anatomy. Screening results for the spinocerebellar ataxia gene were also negative. We thus proposed to expand the scope of genetic screening to include over 200 mutations that can cause ataxia. A final diagnosis of GSS was presented and the patient was followed for more than 3.5 years, during which we noted imaging abnormalities. The patient gradually exhibited decorticate posturing and convulsions. We recommended administration of oral sodium valproate, which resolved the convulsions.CONCLUSION Patients with inherited ataxia should be considered for a diagnosis of GSS via genetic testing at an early disease stage.
Key words: Prion disease; Cerebellar ataxia; Magnetic resonance imaging; Diagnosis;Brain; Case report
INTRODUCTION
Gerstmann-Str?ussler-Scheinker (GSS) disease is a genetic degenerative disease of the central nervous system. It is caused by a mutation in the prion protein gene (PRNP),located on chromosome 20, resulting in the conversion of the encoded prion protein(PrPc), which normally diffuses across cell membranes in the healthy human central nervous system, into insoluble prion protein (PrPsc)[1]. GSS is associated with familial Creutzfeldt-Jakob disease (CJD) and familial fatal insomnia (FFI), which are collectively referred to as human hereditary prion diseases and account for 5%-15% of all prion diseases[2]. GSS disease has an autosomal dominant inheritance pattern and an incidence of only 1-10/108cases annually[3]. The early stages of this disease are primarily characterized by progressive cerebellar ataxia accompanied by pyramidal tract signs and extrapyramidal injury, with severe dementia sometimes occurring later in the disease progression (typically between ages 40-60)[4,5].
In 1936, the neuropathologists Gerstmann, Str?ussler, and Scheinker first reported an Austrian GSS pedigree and coined the disease in their names[6]. In 1991,Kretzschmar et al[7]detected a P102L mutation in the PRNP gene.
The first case of GSS disease in China was diagnosed in 1993 by Professor Lin Shihe[8]using biopsied brain tissue. The brain tissue pathology associated with GSS disease is characterized by local polycentric amyloid plaque deposition in the cerebral cortex, basal ganglia, and cerebellar cortex; astrogliosis and loss of neurons;neurofibrillary tangles; and tau protein deposition[2,9]. However, in more recent years,most GSS disease cases are diagnosed via genetic sequencing. However, genotype and phenotype heterogeneity have rendered the diagnosis of GSS disease difficult. Thus far, GSS disease in China has only been reported in four articles describing eight families in Shanghai, Taiwan, Guangzhou, and Zhejiang[10-13]. The case reported here is the first case of genetically diagnosed GSS disease in Northeast China and has been followed for more than 3.5 years.
CASE PRESENTATION
Chief complaints
A 48-year-old Chinese man was admitted to our hospital with instability while walking and dysarthria for the last 2.5 years and 10 mo, respectively.
History of present illness
He was born in and continued to live in Jilin province and began to experience gradual instability while walking, which was characterized by bilateral foot-dropping and gradual coldness spreading from the feet to the thighs, without any apparent cause, in August 2012. In April 2014, he experienced dysarthria and coughing while drinking water, for which he again sought treatment at another hospital. However, no abnormalities were detected via spinocerebellar ataxia (SCA) genetic sequencing. The aforementioned symptoms gradually progressed to include involuntary head tremors.The patient was unstable even while sitting and experienced unbearable muscle pain in both lower limbs. In February 2015, he was seen at our hospital.
History of past illness
He denied any history of disease.
Personal and family history
The patient’s mother and older brother also exhibited the symptoms described above.However, detailed information pertaining to his other relatives was not available as they had all passed away (Figure 1). His mother developed symptoms at the age of 51 and exhibited severe muscle atrophy in both lower extremities. She was diagnosed with cerebellar atrophy and was bedridden for 6 mo with mild dementia before her death 5 years later. The patient’s older brother experienced onset of the disease at 36 years of age and died after a 9-year disease duration.
Physical examination upon admission
A physical examination upon the patient’s admission revealed dysarthria and slightly decreased muscle strength in his lower limbs. The lower extremity deep tendon reflexes were not present. Postural and resting tremors were observed in his upper extremities without myoclonus. His performance on the finger-to-nose and heel-toknee tests was poor. Babinski and Chaddock signs were absent. He had a wide-based ataxic gait and was positive for the Romberg sign.
Laboratory examinations
Several routine blood tests were conducted, including those for thyroid function,rheumatoid immune antibodies, and tumor markers, the results of which were all normal. His cerebrospinal fluid (CSF) pressure was normal while CSF protein and immunoglobulin (IgG) levels were slightly elevated. Blood and CSF tests for neuromyelitis optica (NMO)-IgG, aquaporin 4 antibodies (AQP4-Ab), and paraneoplastic antibodies were all negative. In addition, SCA genetic sequencing results were negative.
In light of the patient’s clinical symptoms and family history, we proposed to expand the scope of the genetic screening to include over 200 additional possible mutations that are associated with ataxia. Venous blood was collected for further genetic sequencing and a heterozygous mutation site was found at the second exonic sequence of the PRNP gene: c.305C > T (cytosine to thymine). This mutation results in an amino acid change (p.P102L, valine to leucine) (Figure 2).
Imaging examinations
No abnormalities were observed on routine ultrasound examinations. During the initial 2.5 years of the disease, brain, cervical, thoracic, and lumbar magnetic resonance imaging (MRI) scans and electromyography (EMG) revealed no obvious abnormalities.
FINAL DIAGNOSIS
The patient was diagnosed with GSS disease. We recommended that he undergo a brain biopsy and that his relatives undergo genetic sequencing for similar PRNP gene mutations, but they all refused.
TREATMENT
We provided symptomatic supportive care for the patient, which aimed to relieve postural and resting tremors and muscle pain.
OUTCOME AND FOLLOW-UP
The patient was followed for 3.5 years following his discharge. His symptoms gradually progressed and muscle atrophy and myoclonus of the lower extremities occurred. As his disease progressed, the patient became bedridden and could not care for himself. In July 2017 (5 years after symptom onset), the patient became unconscious, was no longer able to answer questions, and was incontinent.Additionally, convulsions manifested as gaze deviations towards one side, trismus,bilateral upper limb flexion, and bilateral lower limb stiffness. These convulsions lasted for approximately 1 min and occurred one to two times per month.
In January 2018 (5.5 years after symptom onset), we again examined the patient.This physical examination revealed a decorticate state, a bilateral pupil diameter of 2 mm, direct and indirect light reflex sensitivity, no movement of the eyes, bilateral upper limb flexion, bilateral lower limb muscle atrophy, quadriplegia, absent tendon reflexes, and bilateral positive Babinski and Chaddock signs.
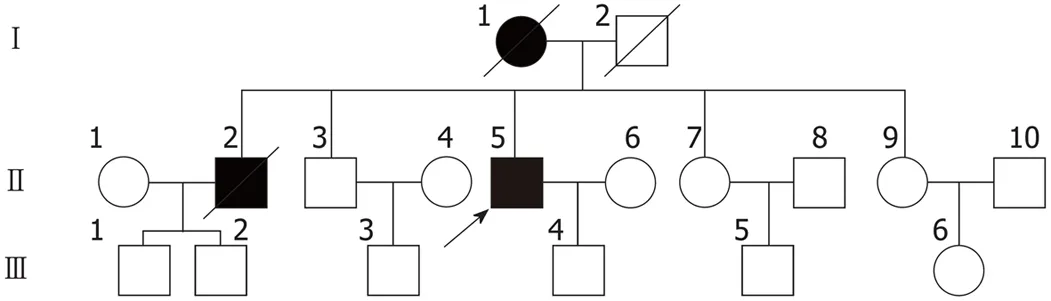
Figure 1 Proband’s pedigree. Squares indicate men, circles indicate women, black symbols indicate affected individuals, diagonal lines across symbols indicate deceased individuals, and the arrow indicates the proband.
Re-examination of brain MRI scans (Figure 3) revealed abnormal signal intensity in the basal ganglia, corona radiata, and the paraventricular and semiovale centers;severe brain atrophy; and ventricular dilation. T1-weighted imaging revealed equal and hypointensive regions while T2-weighted imaging and fluid-attenuated inversion recovery revealed hyperintensive regions. Diffusion weighted imaging (DWI)revealed a diffuse and symmetrical high-intensity signal in the bilateral cerebral cortex while the apparent diffusion coefficient indicated a low-intensity signal. An electroencephalogram (EEG) indicated bilateral frontal, central, and temporal spasmodic mid-waves, tip slow wave intermittent distribution, and irregular waveforms, particularly in the left side of the forehead and palate. Given this imaging signature, we recommended that the patient be administered 0.5 g of oral sodium valproate once daily. Over the subsequent 9-mo follow-up period, the patient experienced no convulsions.
DISCUSSION
The onset of GSS disease typically occurs at the age of 50. It has the longest disease duration among the prion diseases, with an average duration of approximately 5-7 years, although it may continue for up to 12 years[3]. The typical clinical manifestation of GSS disease includes progressive cerebellar ataxia with pyramidal tract signs,pseudobulbar paralysis, myoclonus, and absent tendon reflexes; in many cases,dementia may also occur in the late stages of the disease[4,5]. Brain tissue pathology is predominantly characterized by local polycentric amyloid plaque deposition in the cerebral cortex, cerebellar cortex, and the basal ganglia; loss of neurons and astrogliosis, with or without spongy changes; neurofibrillary tangles; and tau protein deposition[2,9]. Genetic testing has gradually become the most reliable means for diagnosing GSS disease. More than 30 sites of PRNP mutations have been reported, of which P102L (wherein valine is replaced by leucine) is the most common mutation[14].PRNP genotypes and clinical phenotypes are highly heterogeneous with molecular mechanisms depending partially on polymorphisms in the 129thcodon methionine/proline allele.
The patient described in this report, who had a family history positive for GSS disease, experienced ataxia onset with pyramidal tract signs and extrapyramidal manifestations and gradually developed a pseudobulbar palsy. When combined with his gene sequencing results, GSS disease was definitively diagnosed. Brain MRI scans in patients with GSS are typically normal early in the course of the disease, with cortical and/or cerebellar atrophy often appearing with disease progression.
This patient might easily have been misdiagnosed with SCA because he exhibited no brain MRI or EMG examination abnormalities in the 2.5 years following symptom onset. However, recent results[15]have suggested that studies of brain function may have diagnostic implications in the early stages of the disease. The examination of two patients with GSS P102L using99mTc-ethylcysteinate dimer single-photon emission computed tomography (99mTc-ECD SPECT) recently revealed that both exhibited hypothalamic atrophy and reduced blood flow. SPECT in an additional five patients in the early stages of the disease revealed diffuse cerebral blood flow reductions,primarily in the occipital lobe. However, no significant decrease in cerebellar blood flow was observed in these cases, indicating that their lesions predominantly occurred in the brain and spinal cord[15].
Findings such as those described above may be key to distinguishing GSS P102L from other neurodegenerative diseases, such as SCA. In addition, proton magnetic resonance spectroscopy (1H-MRS) has been found to reveal decreased N-acetylaspartate-to-creatine ratios in the cerebellum, frontal corpuscle, and putamen,which may also be helpful in the early diagnosis of GSS disease[16].

Figure 2 Gene mapping of the proband. A heterozygous mutation site was found in the 2nd exonic sequence of the PRNP gene (c.305; cytosine to thymine),resulting in a p.P102L amino acid change (valine to leucine).
Five and a half years after initial symptom onset, their brain MRI scans revealed atrophy and abnormal white matter signals while DWI revealed diffuse and symmetrical high-intensity signals in the bilateral cerebral cortex. These were more extensive than the high-intensity DWI signals previously found in the thalamus,frontal lobe, and occipital lobe. Based on these findings, we speculate that DWI hyperintensity is more sensitive than conventional sequences for the detection of GSS disease.
The pathological changes described above may be related to spongiform changes and astrogliosis. Given that brain MRI scans in patients with GSS have revealed cerebellar atrophy at early stages of the disease, care should be taken to differentiate these from lissencephaly and cerebellar hypoplasias caused by mutations in the LIS1,DCX, RELN, TUBA1A, and VLDLR genes. Lissencephaly and cerebellar hypoplasias may result from deficits in the connections between Purkinje and granule cells which cause subsequent granule cell apoptosis[17]. Resultant MRI imaging in these cases may range from cerebellar midline hypoplasia to gross volume reductions[17]. Thus, it is critical that patient symptoms and EEG results are incorporated in the assessment of these patients. For instance, EEG patterns indicative of epileptic seizures may indicate antiepileptic drug administration, which was found to be effective for the symptom treatment in the patient presented here.
This patient experienced typical cerebellar ataxia symptoms 2.5 years after first symptom onset and did not develop dementia. He was therefore misdiagnosed with SCA. As all SCA genetic screenings were negative, we expanded the genetic screening range and a final diagnosis of GSS disease was ultimately reached. Similarly, seven cases of PRNP gene mutations were detected in 206 patients without SCA and Friedreich’s ataxia but who experienced ataxia in Italy[18]. This suggests that GSS disease should be considered in patients with ataxia and negative SCA genetic testing.
Due to the high degree of clinical heterogeneity in this clinical population, the diagnosis of GSS disease is often challenging and requires differentiation from many other potential diagnoses. Woulfe et al[19]reported a case of GSS disease with a Q217R mutation whose clinical manifestation included atypical frontotemporal dementia and who was diagnosed with Pick’s disease and cortical basal ganglia degeneration 8 and 10 years after symptom onset, respectively. However, when the patient died 13 years after symptom onset, she was finally diagnosed with GSS disease following pathological and genetic testing. Furthermore, Karmon et al[20]reported a case involving a patient diagnosed with multiple sclerosis who experienced the typical symptoms of ataxia and dysarthria. A variety of treatments were ineffective in this case and his symptoms continued to progress. Gene sequencing was performed 1.5 years later and, when combined with family history, this patient was eventually diagnosed with GSS disease. Webb et al[21]studied nearly 100 cases with P102L mutations in the UK and found that some patients with cognitive impairments,psychotic symptoms, abnormal CSF 14-3-3 protein levels, and abnormal EEG results were misdiagnosed with sporadic CJD. However, a small number of patients who presented with dementia and abnormal brain MRI results with T2hyperintensities in the subcortical deep white matter were misdiagnosed with Binswanger disease.Furthermore, cases with brain MRI scans indicating ventricular and corpus callosum lesions have also been misdiagnosed with multiple sclerosis[20]. Thus, genetic testing provides extremely important clinical evidence for the correct diagnosis of GSS disease.
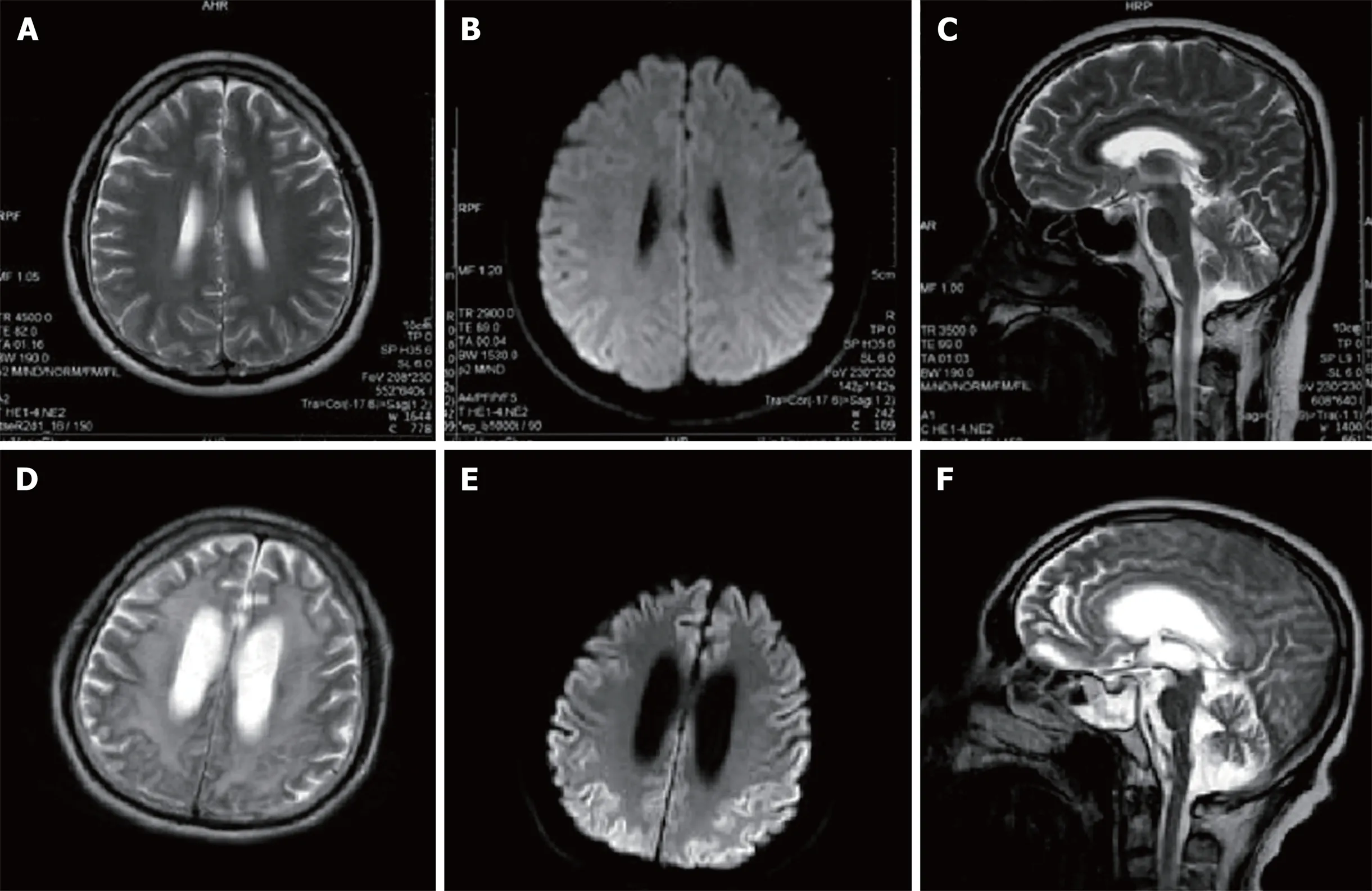
Figure 3 Magnetic resonance imaging scans of the patient’s brain(A, B, D and E: Axial; C and F: Sagittal). A, B, and C: Brain magnetic resonance images were obtained 2.5 years after symptom onset. There are no obviously abnormal signals in the T2-weighted images (A, B) or diffusion weighted image (DWI) (C); D, E and F:Brain magnetic resonance images were obtained 5.5 years after the onset of symptoms. T2-weighted images (D, F) reveal high signal intensities in the basal ganglia,corona radiata, and paraventricular and semiovale centers; severe brain atrophy; and ventricular dilation. DWI imaging (E) reveals a diffuse symmetrical high signal in the bilateral cerebral cortex.
CONCLUSION
Based on the case reported above, we emphasize here that a diagnosis of GSS disease should be considered for patients with inherited ataxia, especially those lacking MRI abnormalities and SCA-related genetic mutations. Given that GSS disease is extremely rare, the pathogenic PRNP gene has many mutation sites and a diversity of associated clinical phenotypes. Attention should thus be focused on diagnosing and treating related clinical symptoms. For instance, early application of SPECT and MRS may indicate GSS disease while genetic testing plays an important role in the diagnosis of GSS disease. We will continue to follow this case, which is the first case of GSS disease in Northeast China, to achieve a more in-depth understanding of the disease and to develop further suggestions for its clinical treatment and avenues for future scientific investigation.
 World Journal of Clinical Cases2019年3期
World Journal of Clinical Cases2019年3期
- World Journal of Clinical Cases的其它文章
- Biventricular pacing for treating heart failure in children: A case report and review of the literature
- Considerations for routine coagulation monitoring with rivaroxaban:A case report and review of the literature
- Castleman disease presenting with jaundice: A case report and review of literature
- Papillary cystadenoma of the parotid gland: A case report
- Ghost cell odontogenic carcinoma of the jaws: Report of two cases and a literature review
- Intravenous leiomyomatosis with different surgical approaches:Three case reports