食用明膠中微量鉻含量測定的方法研究
2019-02-20 12:21:44
食品研究與開發 2019年5期
(廣西-東盟食品藥品安全檢驗檢測中心,廣西南寧530021)
鉻(chromium)廣泛存在于自然界中,人體中微量鉻對人的糖代謝、脂肪代謝、蛋白質及核酸的相互作用有著重要的影響,然而鉻的過度攝入會對人體造成危害[1],國際癌癥研究機構(international agency for researchoncancer,IARC)已將六價鉻列為人類致癌物[2]。食用明膠(gelatin)是用新鮮動物皮骨提取的膠原蛋白質[3],為淡無色至淡黃色薄片狀或粉粒狀末,無臭、無味,應用于食品工業作賦形劑、增稠劑,是食品工業廣泛應用的添加劑。人體如果攝入含鉻明膠,其中的六價鉻會引起細胞周期調控系統CDK6異常表達改變細胞周期和DNA相關修復過程從而引發腫瘤[4]。因此食用明膠鉻含量檢測已成為食品藥品檢測的一個重要指標。目前測定食用明膠中微量鉻的前處理方式主要有微波消解、濕法消解、干法灰化[5];測定方法主要有原子熒光(atomic fluorescence spectrometry,AFS)法、原子吸收-石墨爐(graphite furnace atomic absorption apectrometry,GF-AAS)法和電感耦合等離子質譜(inductively coupled plasma mass spectrometry,ICP-MS)法[6-9]。目前國內相關標準如:GB 5009.123-2014《食品安全國家標準食品中鉻的測定》[10]及GB 5009.268-2016《食品安全國家標準食品中多元素的測定》[11]中已有關于鉻測定的方法,但由于食品基質的復雜性和鉻元素的化學特性,按標準操作會出現某些基質的樣品消解不徹底、儀器測定存在特殊干擾等情況,導致鉻測定的精密度低、準確度差。本研究擬采用3種樣品前處理方法及2種檢測儀器進行綜合全面的比較,以期確立一種快速、便捷、準確,適用于食用明膠中微量鉻的測定方法。
1 材料與方法
1.1 儀器與設備
900T原子吸收光譜儀:PerkinElmer公司;Icap Q電感耦合等離子體質譜儀:Thermo公司;Mars6微波消解儀:CEM公司;P330馬弗爐:Nabertherm公司;BHW-09C石墨電熱消解儀:北京博通公司;ML204電子天平(感量0.1 mg和1 mg):Mettler Toledo公司;Milli-Q超純水儀:Millipore公司。
1.2 材料與試劑
食用明膠質控樣:編號T01530;鉻標準值范圍:380 μg/kg~719 μg/kg,中值:600 μg/kg,分析實驗室能力驗證 (food analysis performance assessment scheme,FAPAS)考核組織提供。
鉻標準溶液、鈧標準溶液:1 000 mg/L,介質為2%硝酸,美國O2SI公司;硝酸、氫氟酸:分析純,德國Merck公司;30%過氧化氫:優級純,廣東光華科技股份有限公司;氬氣:純度99.999%,廣西國信氣體研究有限公司。
1.3 試驗方法
1.3.1 標準溶液配制
1)鉻標準使用液:將鉻標準儲備液用硝酸溶液(5+95)逐級稀釋至濃度為100 ng/mL。
2)鈧標準使用液:將鈧標準儲備液用硝酸溶液(5+95)逐級稀釋至濃度為10 ng/mL。
3)標準系列溶液的配制:分別吸取鉻標準使用液(100 ng/mL)0、0.5、1.00、2.00、3.00、4.00 mL 于 25 mL容量瓶中,用硝酸溶液(5+95)稀釋至刻度,混勻。各容量瓶中每毫升分別含鉻 0、2.00、4.00、8.00、16.0 ng。由GF-AAS和ICP-MS分別吸取標準系列溶液進行測定。
1.3.2 樣品前處理方法
1.3.2.1 微波消解
準確稱取試樣約0.5 g(精確至0.001 g)于微波消解罐中,分別加入6 mL硝酸、4 mL硝酸和2 mL氫氟酸、4 mL硝酸和2 mL雙氧水,按照微波消解的操作步驟消解試樣(消解條件參見表1)。冷卻后取出消解罐,在電熱板上于140℃~160℃趕酸至0.5 mL~1.0 mL。消解罐放冷后,將消化液轉移至10 mL容量瓶中,用少量水洗滌消解罐2次~3次,合并洗滌液,用水定容至刻度。同時做試劑空白試驗、加標回收試驗和平行性試驗,微波消解程序見表1。

表1 微波消解儀工作條件Table 1 Working parameters of microwave digestion
1.3.2.2 濕法消解
準確稱取試樣約0.5 g(精確至0.001 g)于消化管中,分別加入6 mL硝酸、4 mL硝酸和2 mL氫氟酸、4 mL硝酸和2 mL雙氧水,在可調式電熱爐上消解(升溫程序:120℃保持0.5 h、升溫至180℃2 h、升溫至200℃)。若消化液呈棕褐色,再加硝酸,消解至冒白煙,消化液呈無色透明或略帶黃色,取出消化管,冷卻后用水定容至10 mL。同時做試劑空白試驗、加標回收試驗和平行性試驗。
1.3.2.3 干法灰化
準確稱取試樣約2 g(精確至0.001 g)于30 mL坩堝中,置于馬弗爐中,于550℃恒溫3 h~4 h,至試樣呈白灰狀,取出冷卻,用硝酸溶液(1+1)溶解并用水定容至10 mL。同時做試劑空白試驗、加標回收試驗和平行性試驗。
1.3.3 測定方法
1.3.3.1 原子吸收-石墨爐法(GF-AAS)
鉻元素原子化溫度高,經對比,食用明膠的測定即使不加基體改進劑,原子化過程中也不會出現干擾,因此選擇不使用基體改進劑以獲得更高的靈敏度。測定波長:357.87 nm;狹縫寬度:0.7 nm;燈電流:5 mA;進樣體積:20 μL;背景扣除:塞曼扣背景;GFAAS工作參數見表2。
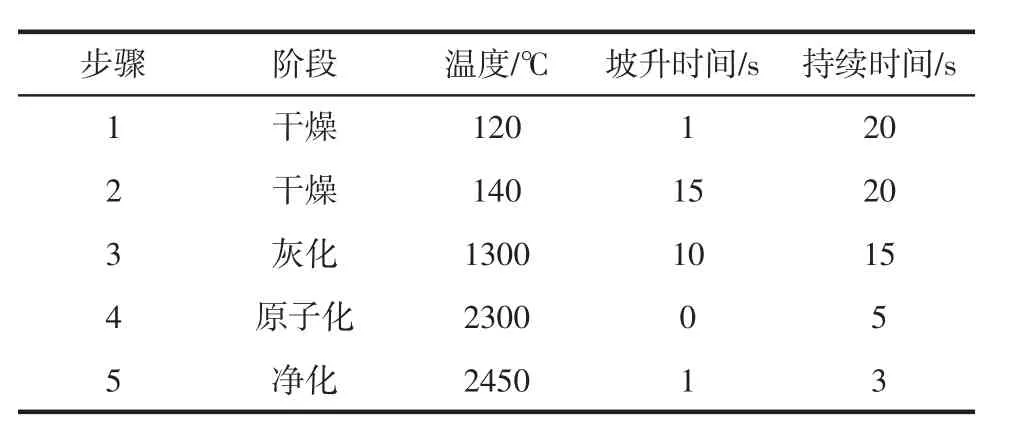
表2 GF-AAS主要工作參數Table 2 Main operating parameters of GF-AAS
1.3.3.2 電感耦合等離子體質譜法(ICP-MS)
鉻同位素有50Cr、52Cr、53Cr、54Cr,其中52Cr天然豐度最高且干擾較低,選為待測同位素。52Cr主要的多原子離子干擾為40Ar12C+、36S16O+、38Ar14N+、36Ar16O+,使用動能歧視型碰撞池(kinetic energy discrimination,KED)模式,通入氦氣作為碰撞氣,能有效減少干擾。ICP-MS測量過程中,被測元素的信號受樣品溶液中共存元素或基體元素的影響,引起被測物信號的增強或減弱的現象,稱之為抑制或增敏效應[12]。抑制或增強效應可通過內標法進行校正,使用10 ng/mL的鈧(Sc)作為內標元素可以減少這些干擾及儀器各種條件的波動帶來的影響,ICP-MS工作參數見表3。
1.3.3.3 鉻含量計算公式
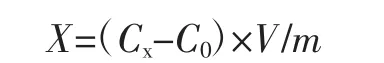
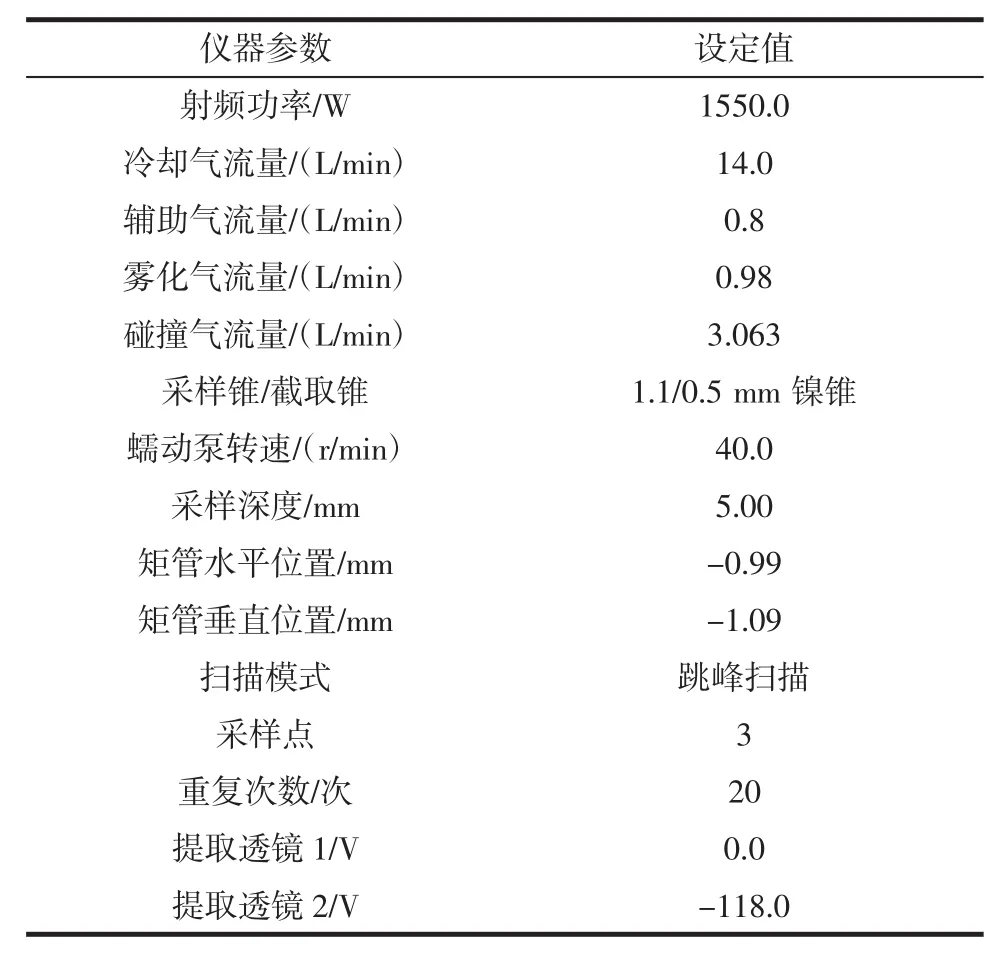
表3 KED模式ICP-MS主要工作參數Table 3 Main operating parameters of ICP-MS in KED mode
式中:X為樣品中鉻的含量,μg/kg;Cx為樣品溶液中鉻的質量濃度,ng/mL;C0為空白溶液中鉻的質量濃度,ng/mL;V為樣品溶液定容體積,mL;m為樣品稱樣量,g。
2 結果與分析
2.1 不確定度分量來源的分析
GF-AAS法及ICP-MS法測定明膠鉻含量的不確定度的主要來自:標準曲線擬合計算濃度過程;樣品重復性測定次數;樣品的處理過程;標準溶液的配制過程[13];分析儀器的相對不確定度。
2.2 樣品測定結果分析
本文比較了3種不同的消解方式,結果見表4和表5。
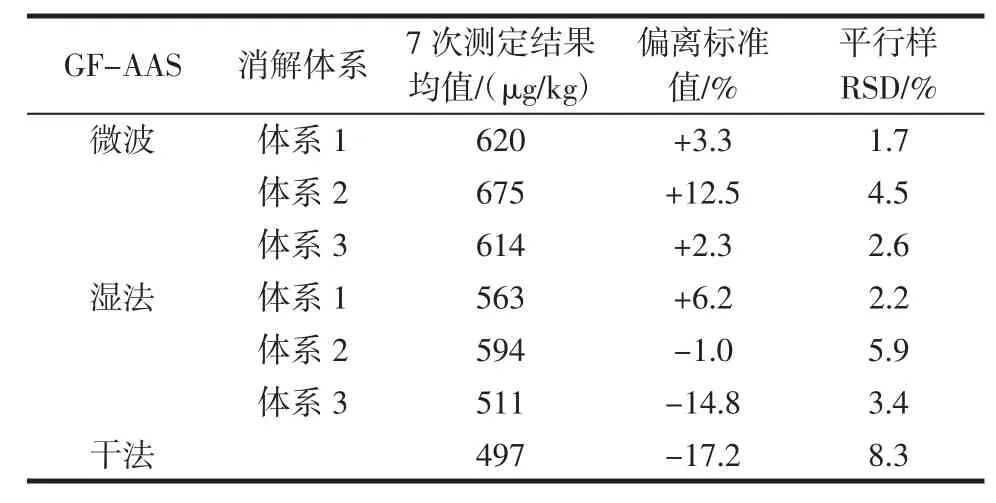
表4 GF-AAS測定鉻結果Table 4 GF-AAS results of chromium
由表4和表5可知,干法灰化法在樣品經馬弗爐灰化后加酸溶解并轉移的過程中,易造成鉻元素損失;濕法消解耗時長且消解不夠徹底;微波消解耗時短且消解效果好,平行樣精密度高。研究了不同的酸體系的消解效果,結果表明,硝酸-過氧化氫體系消解效果最佳,過氧化氫能夠使樣品消解液更澄清透亮,起到氧化脫色作用,羥自由基同時也更利于有機物的分解,從結果來看該消解體系得到的樣品溶液測定結果精密度最高,也接近質控樣的標準值;而硝酸-氫氟酸體系消解過程中引入氟離子,在趕酸過程中即使加入高氯酸也較難將氟趕盡,在高氯酸趕F的操作中,時間不易控制,時間不夠或過長均會導致鉻測定結果的偏高或偏低[14],效果不理想。此外,還比較了不同的儀器,對同一消解方式的測定結果:鉻是高溫元素,在原子化溫度(橫向加熱)高達2 300℃時,干擾極少,原子吸收-石墨爐測定的結果比較穩定,而電感耦合等離子體質譜測定鉻時,鉻易被同質荷比的多原子離子干擾,開啟碰撞模式消除這類干擾則使靈敏度下降,導致測定結果穩定性較原子吸收-石墨爐法差。
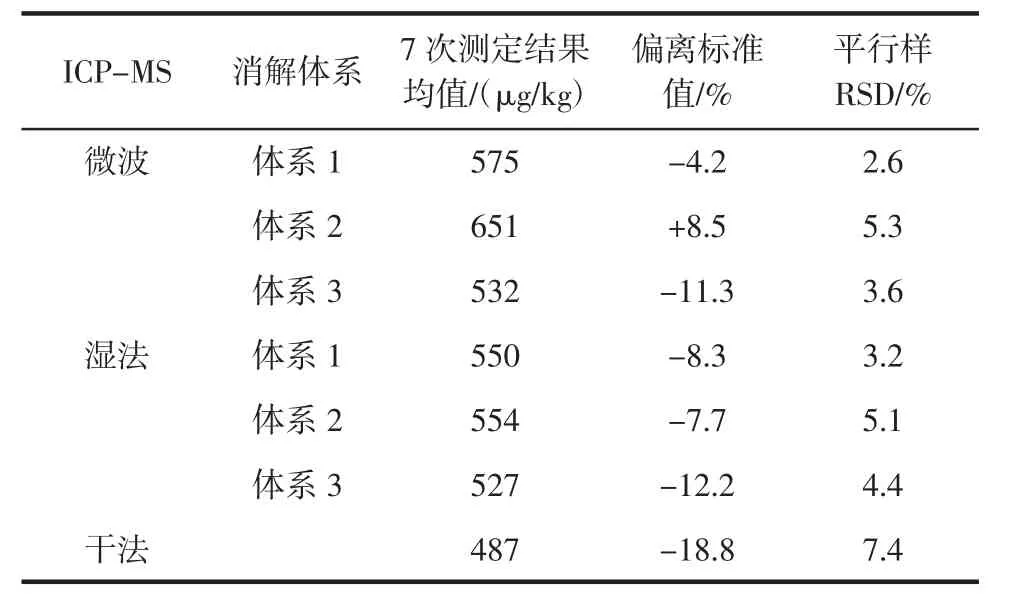
表5 ICP-MS測定鉻結果Table 5 ICP-MS results of chromium
2.3 標準曲線測定結果
鉻標準曲線測定結果見圖1,圖2。
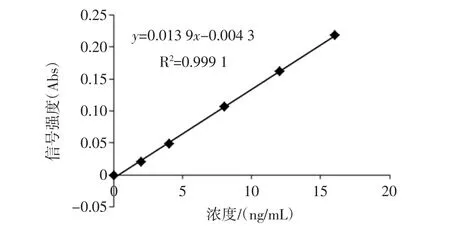
圖1 GF-AAS標準曲線圖Fig.1 GF-AAS calibration curve
2.4 檢出限
GF-AAS和ICP-MS的儀器檢出限均采用純水連續11次測定鉻信號值的3倍標準偏差所相當的鉻濃度,再經過稀釋倍數和取樣質量及單位轉換系數的換算,得到對應方法檢出限GF-AAS法:0.01 mg/kg;ICPMS法:0.006 mg/kg,均能很好地滿足試驗靈敏度需求。

圖2 ICP-MS標準曲線圖Fig.2 ICP-MS calibration curve
2.5 回收率試驗
按照不同前處理方法分別稱取試樣,向其中加入與稱取試樣鉻含量接近的鉻標準溶液,分別按照相應的前處理方法制備得到回收率試樣待測液,供GFAAS及ICP-MS測定,得到不同前處理條件下的試樣回收率,回收率見表6、表7。
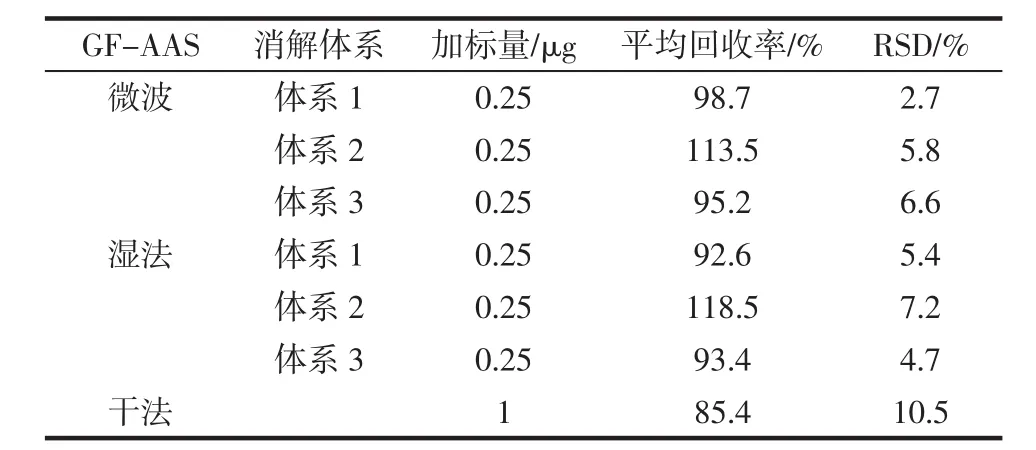
表6 GF-AAS加標回收率Table 6 GF-AAS spike recovery
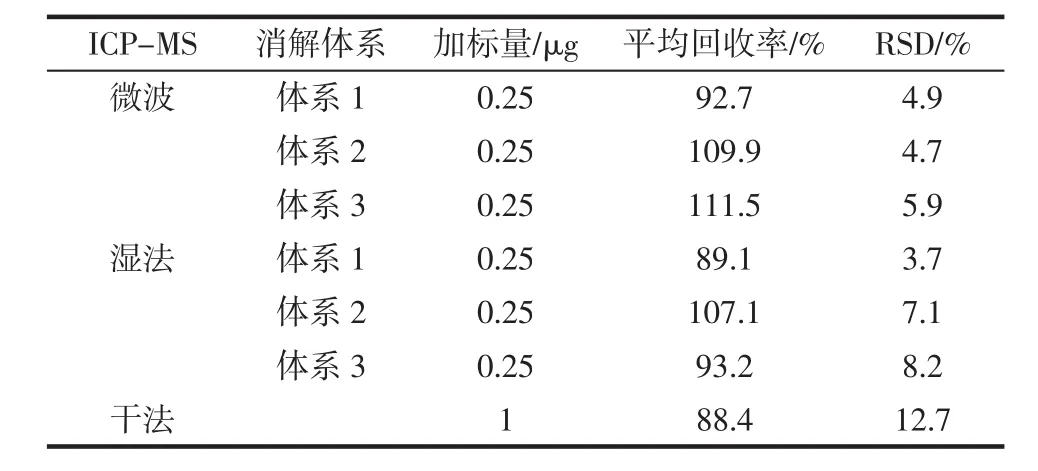
表7 ICP-MS加標回收率Table 7 ICP-MS spike recovery
綜合比較表6、表7中回收率情況,可以看出干法回收率略低,各種酸體系的微波消解和濕法消解所得回收率測定液,在GF-AAS和ICP-MS中測得回收率結果均比較滿意。
2.6 精密度試驗
按照2.2中微波消解(體系1~體系3)、濕法消解(體系1~體系3)、干法灰化的7種前處理方式,各取7份平行樣進行前處理,所得7組待測樣品溶液分別在GF-AAS和ICP-MS上進行精密度測定,結果表明相比較于其他前處理方法,硝酸-過氧化氫體系—微波消解這一前處理方法所得的結果精密度最好,在GFAAS上測得RSD=1.7%,在ICP-MS上測得RSD=2.6%,試驗數據詳見表8、表9。
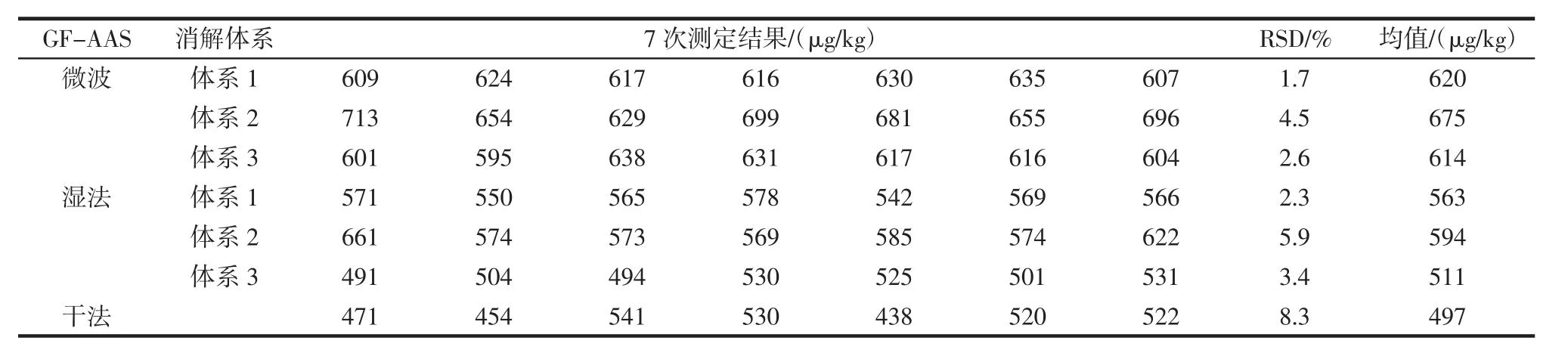
表8 GF-AAS精密度(n=7)Table 8 GF-AAS precision(n=7)
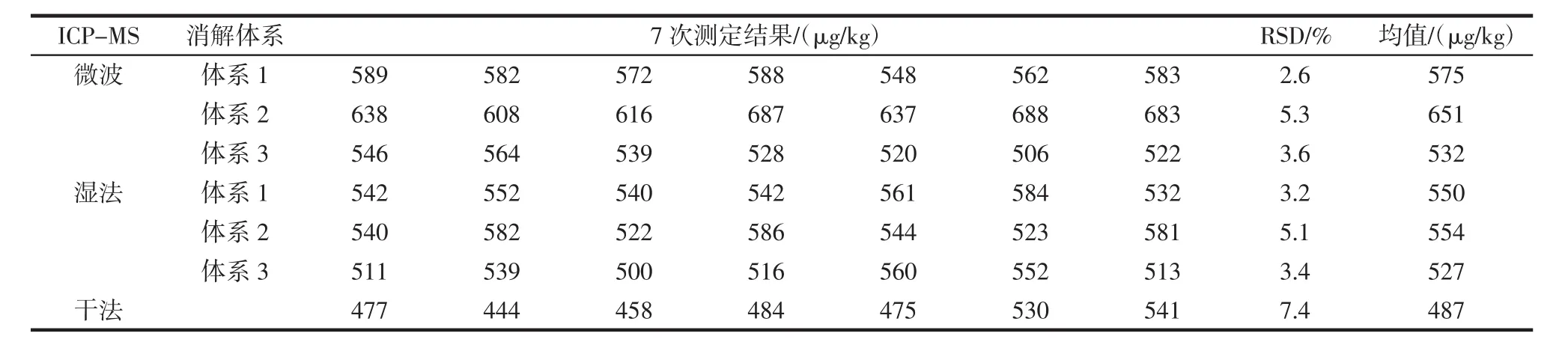
表9 ICP-MS精密度(n=7)Table 9 ICP-MS precision(n=7)
2.7 重現性
取同一食用明膠質控樣,在不同的實驗室設備條件下,由不同的人員按照同樣的方法操作(試驗2),得到的鉻測定結果與原測定結果(試驗1)對比見表10、表11。
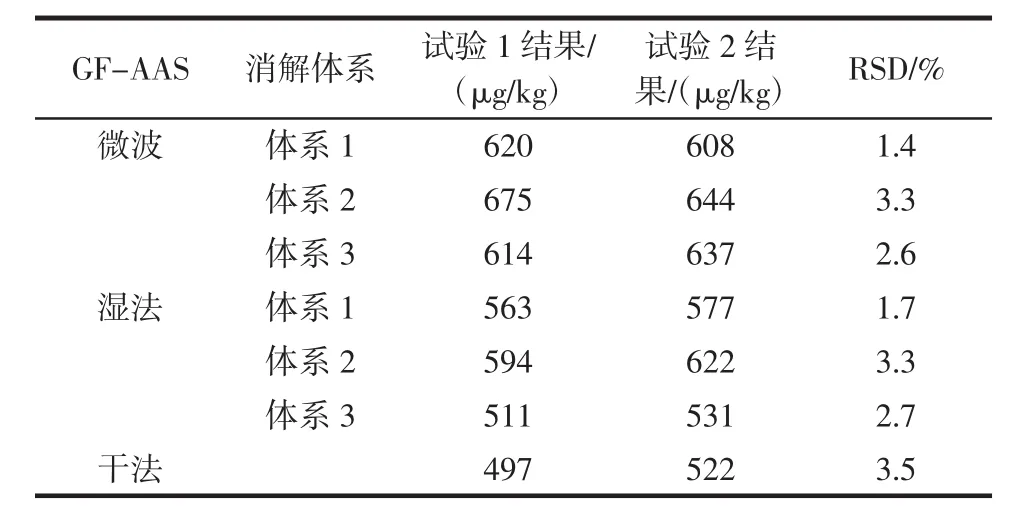
表10 GF-AAS測定鉻結果Table 10 GF-AAS results of chromium
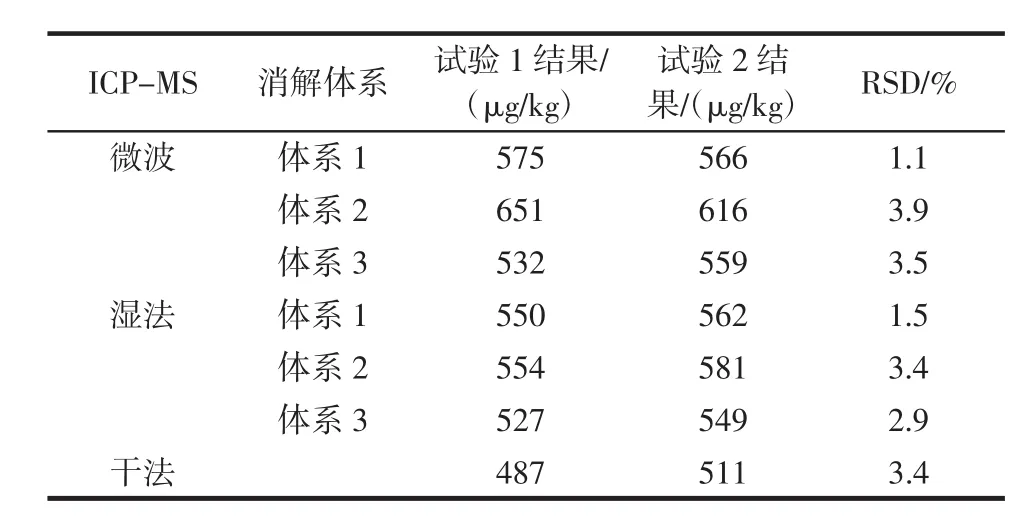
表11 ICP-MS測定鉻結果Table 11 ICP-MS results of chromium
3 結論與討論
硝酸-過氧化氫體系-微波消解-原子吸收-石墨爐這一測定方法,樣品消解效果最好,平行樣測定結果精密度最高,所得結果較接近標準物質鉻含量的標準值,加標回收測定結果最為滿意。如不加過氧化氫溶液,從濕法消解結果及ICP-MS測定結果看來,精密度、偏離標準值、加標回收率等方面結果較差。
本研究經過多方面對比,樣品使用硝酸-雙氧水消解體系進行微波密閉消解并采用原子吸收-石墨爐法測定食用明膠中微量鉻,靈敏度高、精密度高、準確度好、重現性好、回收率高,質控樣測定結果接近其中值,是適合用于明膠中微量鉻測定的較好的方法。