微陣列比較基因組雜交技術在出生缺陷新生兒中的應用研究
2019-02-10 10:48:24林薔楊秀芳鄭鎧軍
川北醫(yī)學院學報 2019年6期
關鍵詞:檢測
林薔,楊秀芳,鄭鎧軍
(中山大學附屬中山市人民醫(yī)院新生兒科,廣東 中山 528400)
據(jù)統(tǒng)計顯示,我國出生缺陷發(fā)生率約為5.6%,每年新增出生缺陷患兒將近90萬例[1],而在出生缺陷患兒中有近30%在5歲前死亡,40%則終生殘疾[2]。生長發(fā)育遲緩(developmental delay,DD)是較為常見的出生缺陷疾病,而導致DD發(fā)生的病因復雜,不明原因的情況占30%~60%[3]。染色體核型分析現(xiàn)已廣泛應用染色體異常的檢測中,但對于5 Mb以下的染色體異常無法準確識別。多重鏈接探針擴增技術可檢測出5%~10%的基因拷貝數(shù)變異情況,但無法檢測基因中的點突變和染色體平衡易位。微陣列比較基因組雜交技術(array-based comparative genomic hybridization,array-CGH)相比傳統(tǒng)細胞遺傳學方法,具有高通量、高分辨率、高敏感性、高準確率等優(yōu)點[4]。本研究采用微陣列比較基因組雜交技術評估DD發(fā)生的遺傳病因,望為其臨床診治和遺傳咨詢提供參考。
1 資料與方法
1.1 一般資料
選取2016年3月至2018年10月中山大學附屬中山市人民醫(yī)院收治的不明原因的DD患兒20例為研究對象,所有患兒均符合DD診斷和篩選標準[5-6]。患兒年齡1~25 d,平均(9.2±6.1)d;男性12例,女性8例。排除已知明確病因者,如遺傳代謝性疾病、顱內(nèi)腫瘤、缺血缺氧性腦病等。所有患兒采用自身對照法,先經(jīng)常規(guī)G顯帶染色體核型分析,設為對照組;再采用微陣列比較基因組雜交技術進一步分析(array-CGH組)。
1.2 方法
1.2.1 DNA提取 采集患兒外周血2 mL,加入乙二胺四乙酸抗凝劑,采用Blood Genome DNA Extraction Kit (takara 公司)提取基因組DNA,總量50 μL以上,濃度100~150 ng/μL,-20 ℃保存待檢。
1.2.2 基因檢測 采用Affymetrix公司配套檢測試劑盒及優(yōu)化的標準操作流程,使用CytoScanHD/CytoScan750K進行全基因組范圍掃描。檢測儀器:AffymetrixGeneChip System 3000Dx v.2芯片系統(tǒng); 檢測芯片:CytoScan HD,包括270萬個探針(195萬個拷貝數(shù)探針和75萬個SNP 探針),除加密覆蓋拷貝數(shù)多變區(qū)、遺傳疾病關聯(lián)區(qū)、基因分布區(qū)等,還均勻分布人類基因組整個范圍。分析軟件:Affymetrix Chromosome Analysis Suite Software;version3.0。質(zhì)量控制:所有實驗在質(zhì)控標準通過的情況下,用Affymetrix Chromosome Analysis Suite Software進行分析,以Affymetrix提供的正常人DNA作為對照標準。
1.3 統(tǒng)計學分析

2 結(jié)果
2.1 患兒臨床特征
20例患兒中,有5例(25%)存在特殊面容(包括頭型窄長,耳朵突出,尖下巴,眼瞼下垂,顱骨畸形,眼距寬,寬鼻梁,v型嘴,小耳等);6例(30%)出現(xiàn)矮小、小陰莖、皮膚缺失、天性心臟病、趾畸形、陰道閉鎖等。見表1。

表1 患兒臨床特征
2.2 兩種檢測結(jié)果比較
20例患兒經(jīng)常規(guī)G顯帶染色體核型分析表明,有2例存在染色體拷貝數(shù)變異,array-CGH檢測發(fā)現(xiàn)有5例存在染色體拷貝數(shù)變異,兩組比較,差異無統(tǒng)計學意義(P>0.05);但兩組病因確診率比較,差異有統(tǒng)計學意義(P<0.05)。見表2。
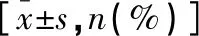
表2 兩種檢測結(jié)果比較
2.3 array-CGH檢測結(jié)果
20例患兒中,有5例(25%)存在染色體拷貝數(shù)變異,根據(jù)現(xiàn)有的文獻和數(shù)據(jù)庫資料結(jié)合array-CGH檢測報告,4例患兒明確病因,其中1號患兒在chr22q13.31q13.33區(qū)域發(fā)生3.8 Mb片段缺失,為22q13.3缺失綜合征,又被稱為Phelan-McDermid綜合征;2號患兒在chr15q11.2q13.1區(qū)域發(fā)生約6.2 Mb片段缺失,與Prader-Willi綜合征相關;3號患兒在chr11q24.2q25區(qū)域發(fā)生10.3 Mb片段缺失,為Jacobsen綜合征;4號患兒在chr5q32處存在約311 Kb缺失片段,該片段缺失可致Treacher Collins綜合征 ;5號患兒多條染色體發(fā)生了大片段純合子(>3 Mb)現(xiàn)象,片段長度總和大約為171.0 Mb,占常染色體基因組片段總長度的6.0%,臨床意義尚不明確。 其余患兒染色體拷貝數(shù)為正常多態(tài)性改變。見表3、圖1、圖2、圖3、圖4和圖5。

表3 20例患兒array-CGH檢測結(jié)果


2.4 染色體拷貝數(shù)變異情況
5例患兒存在染色體拷貝數(shù)變異,其中1號患兒基因組突變位點及涉及基因為arr[hg19] 22q13.31q13.33(47,270,321-51,115,526)x1/SHANK3(606230);2號患兒為arr[hg19] 15q11.2q13.1(22,770,421-28,928,730)x1/UBE3A;3號患兒為arr[hg19] 11q24.2q25(124,630,054-134,938,470) x1;4號患兒為arr[hg19] 5q32(149,477,431-149,788,137) x1/CSF1R (164770),PDGFRB (173410),CAMK2A(114078),TCOF1(606847);5號患兒在多條染色體上發(fā)生大片段純合子(hg19)。見表4。

表4 5例患兒基因拷貝數(shù)變異情況
3 討論
染色體拷貝數(shù)變異在神經(jīng)性發(fā)育障礙,如智力低下、生長發(fā)育遲緩、精神分裂癥、孤獨癥等發(fā)病機制中起重要作用[7]。傳統(tǒng)的G顯帶核型分析技術對染色體不平衡畸變引起的出生缺陷具有較廣泛的應用,但對于5 Mb以下的染色體異常往往無法準確識別[8]。微陣列比較基因組雜交技術通過對全基因組的掃描,突破了傳統(tǒng)細胞遺傳學方法的諸多限制,可檢測出染色體亞顯微缺失和重復突變,具有高通量、高分辨率等優(yōu)點[9-10]。研究[11]指出,在智力低下、多發(fā)性先天性畸形等患者的產(chǎn)后遺傳診斷中,array-CGH檢出率為7%~11%,可檢測出8%的常規(guī)技術無法檢測到染色體失衡。何璽玉等[12]通過array-CGH技術對不明原因ID/DD患兒進行檢測,發(fā)現(xiàn)38%的患兒存在染色體拷貝數(shù)異常,為19%的患兒明確了病因。歐明林等[13]通過探討array-CGH技術在染色體異常細胞遺傳學分析中的作用時指出,array-CGH技術有利于篩查病理性染色體拷貝數(shù)異常,可明確染色體易位類型。本研究發(fā)現(xiàn),20例患兒中,有5例(25%)存在染色體拷貝數(shù)變異,根據(jù)現(xiàn)有的文獻和數(shù)據(jù)庫資料結(jié)合array-CGH檢測報告,4例(20%)患兒明確了病因,與傳統(tǒng)的G顯帶核型分析技術檢測結(jié)果存在顯著差異。說明array-CGH技術在檢測染色體亞顯微缺失和明確病因方面具有較好的應用價值。
此外,array-CGH技術也存在相應的局限性,例如對染色體平衡倒位、易位等染色體結(jié)構異常無法準確識別,僅可對特定分辨率的染色體拷貝數(shù)的缺失和增加進行篩選,無法對更小的染色體異常及基因點突變做出準確的準斷。另外,目前基因芯片價格相對較貴,還未得到更廣泛的應用。但正如上文所述,array-CGH技術相比傳統(tǒng)的G顯帶核型分析技術存在諸多優(yōu)點,如在本研究中,傳統(tǒng)的G顯帶核型分析技術無法確診的患兒,經(jīng)array-CGH檢測明確了病因。如1號患兒經(jīng)array-CGH檢測發(fā)現(xiàn)在chr22q13.31q13.33區(qū)域發(fā)生3.8 Mb片段缺失,涉及SHANK3重要功能基因,為Phelan-McDermid綜合征[14]。該綜合征臨床表現(xiàn)為發(fā)育遲緩、中到重度智力低下、有特殊面容等[15]。1號患兒臨床特征與上述癥狀基本相符,可進一步判斷為Phelan-McDermid綜合征。2號患兒在chr15q11.2q13.1區(qū)域發(fā)生約6.2 Mb片段缺失,與Prader-Willi綜合征相關,該綜合征是一種復雜的遺傳性疾病,嬰兒期主要臨床表現(xiàn)為發(fā)育遲緩,特殊面容等,與患兒臨床特征相符,其發(fā)病率為1/30 000~1/10 000[16]。3號患兒在chr11q24.2q25區(qū)域發(fā)生10.3 Mb片段缺失,為Jacobsen 綜合征[17]。常見的臨床表現(xiàn)包括產(chǎn)前/產(chǎn)后物理性生長遲緩,顯著的面容畸形等,與患兒臨床特征相符。該綜合征15%的病例因家族性平衡易位導致的不平衡分配或其他染色體重排引起[18]。4號患兒在chr5q32處存在約311.0 Kb缺失片段,包含CSF1R,PDGFRB,CAMK2A,TCOF1等功能基因,該片段缺失可致Treacher Collins綜合征。常伴發(fā)育遲緩,顱面發(fā)育異常等,與患兒臨床特征相符。TCOF1功能基因缺失可導致先心病的發(fā)生[19-21]。此外,5號患兒經(jīng)檢測發(fā)現(xiàn)多條染色體發(fā)生大片段純合子(>3 Mb)現(xiàn)象,片段長度總和大約為171.0 Mb,占常染色體基因組片段總長度的6.0%,臨床意義尚不明確,但可能增加了相應區(qū)域內(nèi)一些隱性遺傳病的發(fā)病風險。
綜上,array-CGH技術在檢測染色體亞顯微缺失和明確病因方面具有顯著的診斷價值,值得推廣應用。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:36
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:34
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:50
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:48
