頂空固相微萃取-氣相色譜串聯質譜結合保留指數分析杭白菊揮發性成分
2019-01-29 10:41:32張鵬云李蓉李浩洋龍春霞楊璐齊張峰
食品與發酵工業 2019年1期
張鵬云,李蓉,李浩洋,龍春霞,楊璐齊,張峰
1(廣東藥科大學 公共衛生學院,廣東 中山,528458) 2(中山出入境檢驗檢疫局 檢驗檢疫技術中心,廣東 中山,528400) 3(中國檢驗檢疫科學研究院 食品安全研究所,北京,100176)
菊花(Chrysanthemiflos)是菊科菊屬宿根性草本植物的干燥頭狀花序,根據產地和加工方法主要分為“亳菊”、“滁菊”、“貢菊”、“杭菊”和“懷菊”等[1]。2009年衛生部把菊花加入“藥食同源”物品名單[2]。杭白菊產于浙江桐鄉,在我國有悠久的栽培歷史,具有抗炎、抗腫瘤、抗氧化、保護心血管等多方面的生物活性[3],而揮發油在抗腫瘤、解熱鎮痛、抗病毒、抗炎及促進藥物吸收等方面也具有藥理活性[4],所以菊花中所含的揮發油與其生理活性密切相關。
目前,國內外對揮發油的提取多采用溶劑浸提[5]、水蒸汽蒸餾[6-7]、超臨界CO2流體萃取[8-9]、超聲波輔助萃取[10]、頂空-固相微萃取[11]等方法,再利用氣相色譜質譜串聯技術(gas chromatography-mass spectrometry,GC-MS)檢測其成分。頂空-固相微萃取技術(head space-solid phase micro-extraction,HS-SPME)是近些年發展起來的前處理方法,它集采樣、萃取、濃縮、進樣于一體,操作簡便、使用樣品量少、易實現自動化,大大加快了分析檢測的速度[12]。對揮發性未知物定性分析時,通常根據譜圖檢索的匹配度高低來確認化合物,但是在高匹配化合物較多時,容易出現定性錯誤。而保留指數也是色譜的一個定性參數,在對同分異構體、同系物和結構特征相似的化合物進行定性分析時起到更加重要的作用[13-14]。
本實驗以杭白菊為研究對象,采用頂空固相微萃取-氣相色譜-三重四極桿串聯質譜技術,檢測杭白菊的揮發性成分,對萃取條件進行優化,對檢測出的揮發性成分進行定性分析。利用自動質譜退卷積定性系統(AMDIS)識別拆分共流色譜峰,得到更純凈的質譜圖,再結合Kováts保留指數(retention index,IR)使質譜檢索結果更為準確。
1 材料與方法
1.1 材料與試劑
杭白菊藥材:干花(含水量18.1%),采自浙江桐鄉;氯化鈉:分析純,廣州化學試劑廠;正構烷烴混合標準品(C7~C40),美國O2si公司。
1.2 儀器與設備
TSQ 8000氣相色譜-三重四級桿串聯質聯儀(GC-MS/MS,配電子電離源及Xcalibur數據處理系統)、TRIPLUS RSH自動進樣器、20 mL頂空樣品瓶、固相微萃取自動進樣手柄,美國Thermo公司;固相微萃取頭(65 μm PDMS/DVB、85 μm CAR/PDMS、85μm PA、50/30 μm DVB/CAR/PDMS),美國Supelco公司。
1.3 實驗方法
1.3.1 頂空固相微萃取條件
稱取杭白菊碎片0.3 g,置于20 mL頂空瓶中,加入1.0 g NaCl和8 mL蒸餾水,萃取溫度70 ℃,萃取時間50 min,平衡時間10 min,解吸時間5 min,萃取頭為50/30 μm DVB/CAR/PDMS。
1.3.2 GC-MS/MS分析條件
色譜條件:TR-5MS彈性石英毛細柱(30 m×0.25 mm,0.25 μm);升溫程序:初始溫度40 ℃,保持1 min,然后以13 ℃/min的速率升溫至100 ℃,以3 ℃/min的速率升溫至110 ℃,保持1 min,再以5 ℃/min的速率升溫至150 ℃并保持1 min,以2 ℃/min的速率升溫至180 ℃,保持1 min,最后以15 ℃/min的速率升溫至250 ℃并保持2 min;分流比10:1;進樣口溫度為250 ℃;載氣:99.999% 高純度氦氣;流速1.2 mL/min。
質譜條件:EI離子源;離子源溫度:280 ℃;傳輸線溫度:280 ℃;電子轟擊能量:70 eV;掃描方式:全掃描;質量掃描范圍m/z33~350。
1.3.3 保留指數的測定
采用相同的氣相色譜條件測定正構烷烴(C7~C40)的保留時間,自動質譜退卷積定性系統(AMDIS)根據正構烷烴的保留時間,自動計算出杭白菊中各揮發性化合物的保留指數。保留指數的計算如公式(1):
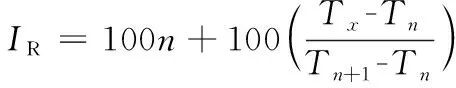
(1)
式中:IR為待測化合物的保留指數;n為正構烷烴的碳原子數;Tx為待測化合物的保留時間,min;Tn和Tn+1分別為碳原子數為n、n+1的正構烷烴的保留時間,min,且Tn+1>Tx>Tn。
1.3.4 數據處理
數據處理由Xcalibur系統軟件完成,應用自動質譜退卷積定性系統(AMDIS)對總離子流圖(TIC)自動進行反卷積處理,所分辨的質譜在NIST2.2標準譜庫中檢索,根據匹配度、保留指數和文獻已報道物質進行核對,只記錄正匹配和反匹配均大于800的物質,并用峰面積歸一化法計算杭白菊揮發油成分的相對含量。
2 結果與分析
2.1 固相微萃取條件的優化
據相關文獻[6,15]報道,杭白菊揮發油的主要成分是萜烯類及其含氧衍生物。因此,本研究用峰面積代表萃取物質的量,出峰數代表萃取物質的種類,選擇杭白菊揮發性成分的總峰面積、主峰面積(萜烯類及其含氧衍生物)、總峰數和主峰數4個參數來評價萃取效果,確定HS-SPME萃取杭白菊揮發性物質的最佳條件。
2.1.1 萃取頭的選擇
稱取杭白菊碎片0.3 g,置于20 mL的頂空樣品瓶中,加入8 mL蒸餾水,萃取溫度為40 ℃,萃取時間為20 min,平衡時間為10 min,分別考察已老化好的不同萃取頭(85 μm CAR/PDMS、65 μm PDMS/DVB、50/30 μm DVB/CAR/PDMS、85 μm PA)對杭白菊揮發性物質的萃取效果,每個樣品重復做3次。結果見圖1。
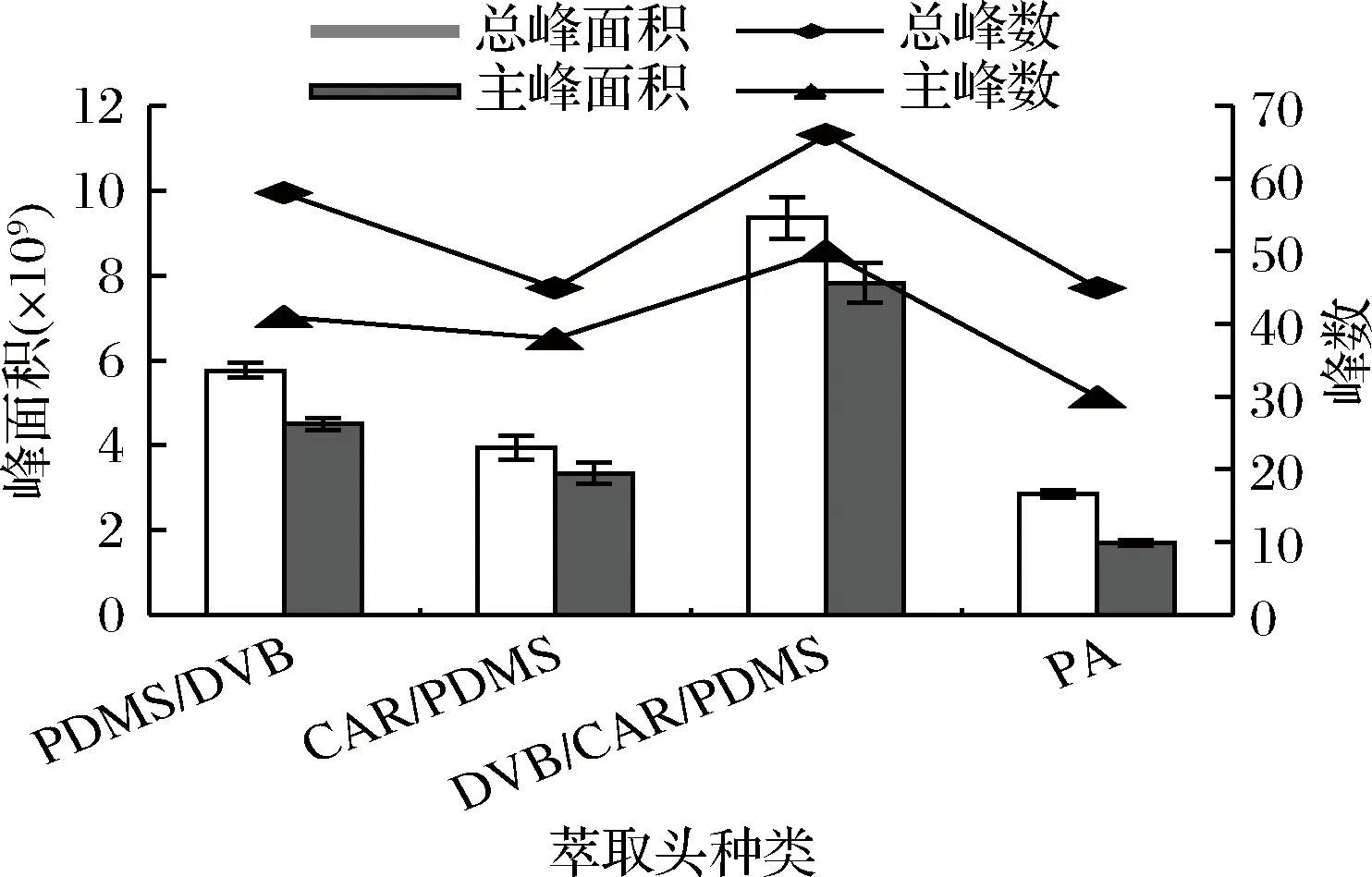
圖1 萃取頭對杭白菊揮發性成分萃取效果的影響
Fig.1 Effect of SPME fiber on the extraction of volatile compounds from Chrysanthemum moriflium Ramat
由圖1可以看出,4種萃取頭的萃取效果有明顯差異。當用50/30 μm DVB/CAR/PDMS萃取頭時,總峰面積、總峰數、主峰面積和主峰數均為最大;當用85 μm PA萃取頭時,峰面積和出峰數均為最小。綜合比較,4種萃取頭對杭白菊揮發性物質的萃取效果依次為DVB/CAR/PDMS >PDMS/DVB>CAR/PDMS>PA。這是因為不同型號的萃取頭具有不同極性的固相涂層,根據相似相溶原理其對化合物的吸附能力也不同[16]。PDMS/DVB萃取頭適用于醇類、醛類、酮類等雙極性化合物; CAR/PDMS萃取頭對于小分子化合物具有較好的吸附能力;DVB/CAR/PDMS萃取頭具有3種涂層,對非極性和混合極性化合物具有較好的吸附性[17];PA是極性萃取頭,適用于極性半揮發性化合物。本研究中杭白菊揮發油成分較復雜,混合涂層更加有利于分析,所以本研究選用50/30 μm DVB/CAR/PDMS。
2.1.2 萃取溫度的選擇
將杭白菊樣品分別在40、50、60、70、80 ℃下,用50/30 μm DVB/CAR/PDMS萃取頭萃取20 min,檢測揮發性成分,每個樣品重復3次。結果顯示,隨著萃取溫度的升高,總峰面積和主峰面積都不斷增加;當溫度超過70 ℃時,雖然總峰面積略有增加,但是主峰面積卻有所減少(見圖2)。從圖2中還可以看到,總峰數和主峰數隨著萃取溫度升高呈先上升后降低趨勢,在70 ℃時達到最大值,說明在高溫下可能有部分不穩定化合物發生分解。據EZQUERRO等[18]報道,萃取溫度對萃取效果影響較大,當溫度升高時,加快了揮發性物質的分子運動速率,使其在氣相中的含量增大,有利于萃取頭對目標物的吸附。但萃取是一個放熱過程,高溫會降低揮發性物質在萃取頭和樣品之間的分配系數,使萃取效果降低[19]。而且高溫下,樣品中的不穩定物質易分解,影響研究結果。綜合考慮各方面因素,本研究將萃取溫度設定為70 ℃。
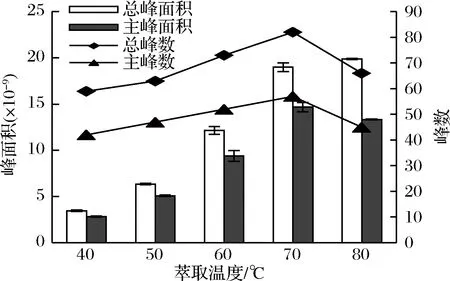
圖2 萃取溫度對杭白菊揮發性成分萃取效果的影響
Fig.2 Effect of extraction temperature on the extraction of volatile compounds from Chrysanthemum moriflium Ramat
2.1.3 萃取時間的選擇
樣品萃取溫度為70 ℃時,用DVB/CAR/PDMS萃取頭分別萃取20、30、40、50 min,檢測揮發性成分,每個樣品重復做3次。結果顯示,當萃取時間在20~40 min之間時,隨著萃取時間的增加,總峰面積和主峰面積都逐漸增大,總峰數和主峰數變化不明顯。當萃取時間超過40 min時,雖然總峰面積和主峰面積變化不大,但是總峰數變化明顯。當萃取時間為50 min時,總峰數和主峰數都達到最大值,而萃取時間為60 min時,總峰數和主峰數卻顯著降低,說明有揮發性成分從萃取頭上解吸出來(見圖3)。不同萃取時間對低分子量揮發性物質和高分子量揮發性物質的峰面積影響不大。萃取頭吸附揮發性化合物是一個動態平衡過程,當分析物在萃取頭、頂空和溶液3相中達到平衡時的萃取效果最好,所需時間即為萃取時間[20]。當萃取時間不足時,揮發性化合物不能充分被萃取頭吸附;而萃取時間過長時,某些化合物可能會從萃取頭上解吸下來[21]。而且,萃取時間過長可能會導致性質不穩定,化合物發生氧化分解等副反應[22]。綜合考慮,本方法將萃取時間設定為50 min。
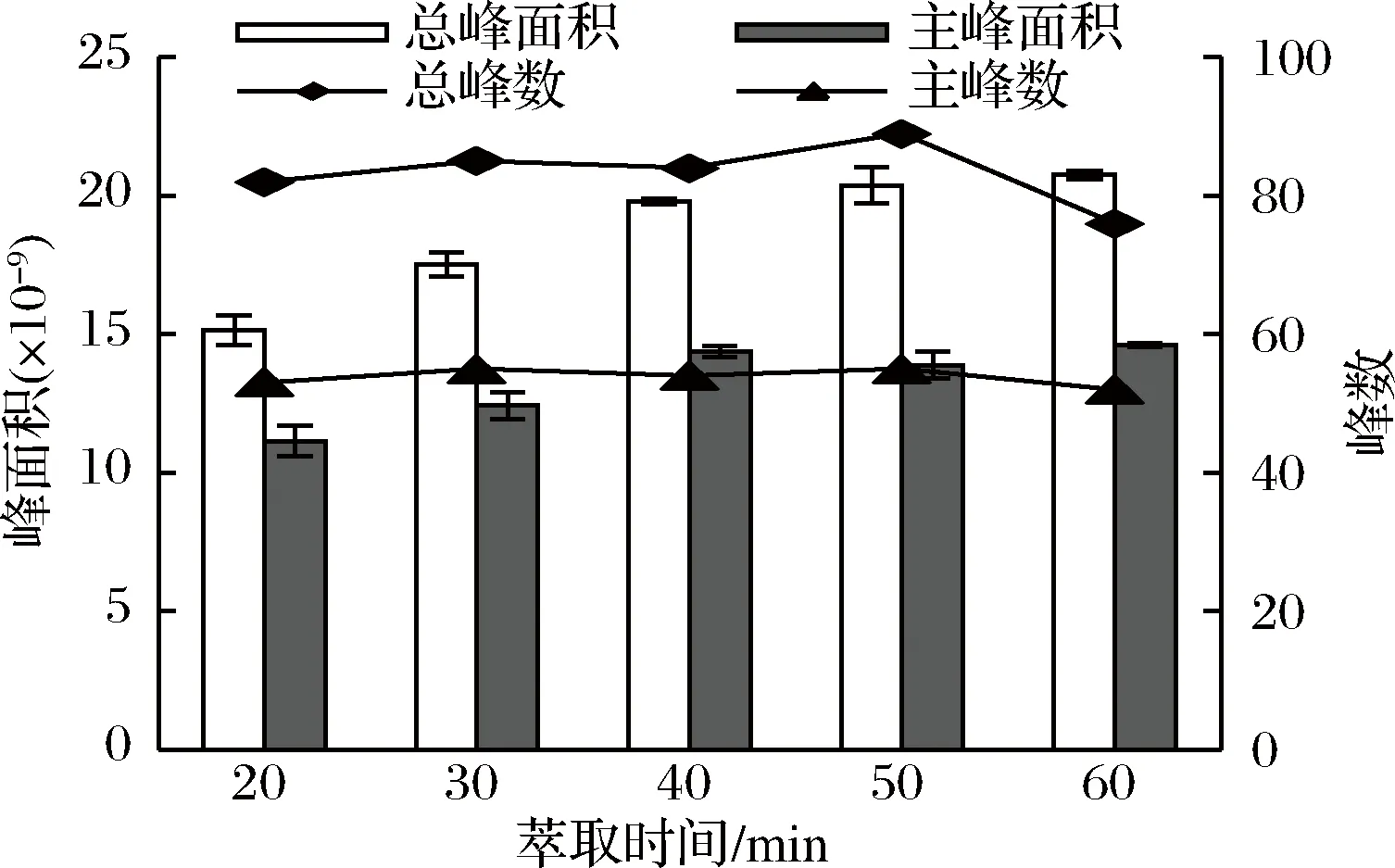
圖3 萃取時間對杭白菊揮發性成分萃取效果的影響
Fig.3 Effect of extraction time on the extraction of volatile compounds from Chrysanthemum moriflium Ramat
2.1.4 氯化鈉(NaCl)的加入量
據LEE[23]、艾對[21]等報道,在樣品中加入氯化鈉可利用鹽析效應降低揮發性成分在水中的溶解度,使揮發性成分充分逸出,尤其利于小分子化合物揮發,增加其在頂空瓶上空的濃度,提高萃取靈敏度。但當NaCl添加過量時,基質黏度增加,影響揮發性成分從水中的逸出,降低萃取效率[24]。實驗將杭白菊樣品分別添加0.0、0.5、1.0、1.5、2.0、2.5、3.0 g NaCl,在70 ℃下用DVB/CAR/PDMS萃取頭萃取50 min,檢測揮發性成分,結果顯示,隨著NaCl添加量的增加,總峰面積、總峰數、主峰面積和主峰數都呈先上升后下降的趨勢,且當添加量為1.0 g時,4個指標都達到最大值。當加鹽量超過2.5 g時,總峰面積、主峰面積和主峰數基本保持不變(見圖4)。說明添加少量NaCl有利于揮發性成分析出,提高萃取效果。因此,本研究選擇加入1.0 g NaCl。
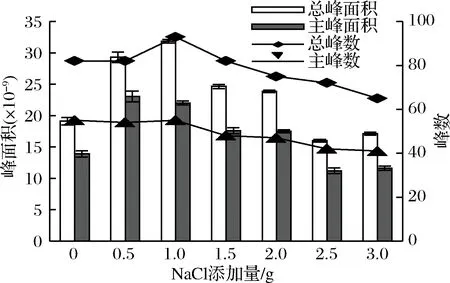
圖4 NaCl添加量對杭白菊揮發性成分萃取效果的影響
Fig.4 Effect of NaCl addition on the extraction of volatile compounds from Chrysanthemum moriflium Ramat
2.1.5 解吸時間的選擇
萃取頭吸附完成后,要在氣相進樣口進行解吸,解吸時間決定著揮發性化合物是否能夠完全進入氣相色譜柱。當解吸時間不足時,揮發性化合物在萃取頭上有殘留,不但影響最終檢測結果,還會影響后續進樣,而當解吸時間過長時,使萃取頭在高溫下遭到損害,影響其壽命,還會使部分物質發生熱解反應,降低檢測效率[25-26]。實驗將杭白菊樣品中添加1.0 g NaCl,在70 ℃下,用DVB/CAR/PDMS萃取頭萃取50 min,然后分別在進樣口解吸3、4、5、6 min,檢測揮發性成分,結果顯示,當解吸時間從3 min增加到4 min時,總峰面積和主峰面積變化不大,總峰數和主峰數都有所增加;而當解吸時間為5 min時,總峰面積急劇增大,總峰數、主峰面積和主峰數均達到最大值,而解吸時間為6 min時,總峰面積又顯著降低,且其余3個參數均為最小值(見圖5)。說明在5 min時,已經解吸完全。因此,本研究將解吸時間設定為5 min。
2.2 實際樣品分析
稱取0.3 g杭白菊碎片于頂空樣品瓶中,加入8 mL蒸餾水,添加1.0 g NaCl,用DVB/CAR/PDMS萃取頭在70 ℃下萃取50 min,解吸5 min來檢測杭白菊中的揮發性成分。通過氣相色譜-三重四級桿串聯質譜儀對分離得到的色譜峰進行定性分析,共鑒定出88個組分,并用峰面積歸一化法得到各組分的相對含量(表1),其總離子流圖見圖6。
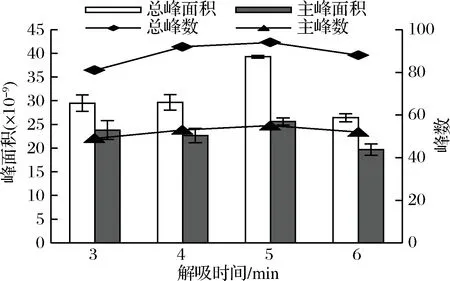
圖5 解吸時間對杭白菊揮發性成分萃取效果的影響
Fig.5 Effect of desorption time on the extraction of volatile compounds from Chrysanthemum moriflium Ramat

表1 杭白菊的揮發性化學成分
續表1

序號揮發性成分匹配度保留指數正匹配反匹配計算值參考值相對含量29壬醛852891110511040.7230苯乙醇945956112011160.0331異佛爾酮85386911271124 0.0632菊油環酮898899113011230.8533菊烯醇878879113911420.9534反式馬鞭草烯醇859862114911440.2435樟腦934934115111451.1636反式-2-壬烯醛881881115911620.0737順式-菊烯醇948949116611620.4138松香芹酮835892117011640.1639龍腦879894117311830.4140(-)-4-萜品醇926926118411821.6441順-3-己烯基丁酯815818118811870.1242對甲基苯乙酮892898119311830.5443萘955956119511820.7644α-松油醇887894119811890.35458,9-脫氫百里酚895921122412210.2446順式-香芹醇910910122512290.3247對枯烯醇926927123312270.2448反-乙酸菊烯酯949951123912390.47492-甲基-3-苯基丙醛898904124812440.0750右旋香芹酮883883125112460.1351順-乙酸菊烯酯948953126612620.6952天竺葵酸856894127012730.2253乙酸龍腦酯933933129212851.9654香芹酚884894129812990.7055β-甲基萘893900130412980.18563,4-二甲基-6-乙基苯酚824845130813050.3457反-乙酸香芹酯928928134113370.1558丁香酚873882136513570.3459順-乙酸香芹酯822894136713620.3160癸酸931933137113730.36612-甲基丁酸苯甲酯866866139213920.9762β-欖香烯927928139813915.9063γ,4-二甲基-4-苯基丁醛804899140914060.5164石竹烯930931142814190.4965(Z)-2-甲基巴豆酸芐酯873896144714460.7366香葉基丙酮895898145414530.2767(E)-β-合歡烯926939145714570.8368倍半香檜烯911923145914640.7169脫氫倍半桉樹腦8919151472147310.95704.5-二-非手性-馬兜鈴烯913920147714693.0671γ-姜黃烯831832148214800.2872α-姜黃烯911940148614833.4373朱欒倍半萜859871149214922.1774佛術烯895897149614992.8475順-β-愈創木烯846881150414992.3176β-沒藥烯841857151215090.2877β-倍半水芹烯904929152715242.2778石竹烯氧化物794818158315811.5879aR-姜黃醇816827158515831.1480十氫二甲基甲乙烯基萘酚800809166516601.6181β-紅沒藥醇858862167516711.52
續表1
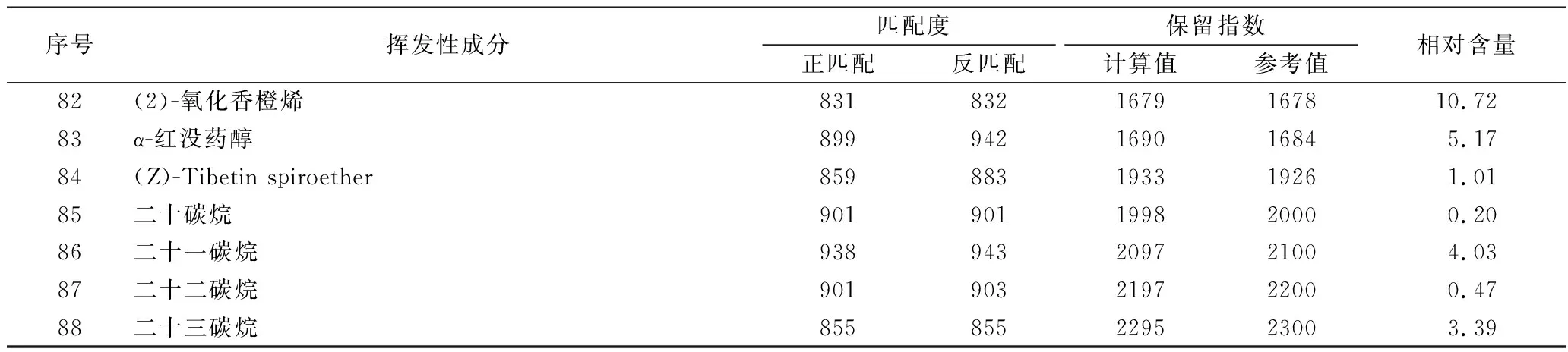
序號揮發性成分匹配度保留指數正匹配反匹配計算值參考值相對含量82(2)-氧化香橙烯8318321679167810.7283α-紅沒藥醇899942169016845.1784(Z)-Tibetin spiroether859883193319261.0185二十碳烷901901199820000.2086二十一碳烷938943209721004.0387二十二碳烷901903219722000.4788二十三碳烷855855229523003.39
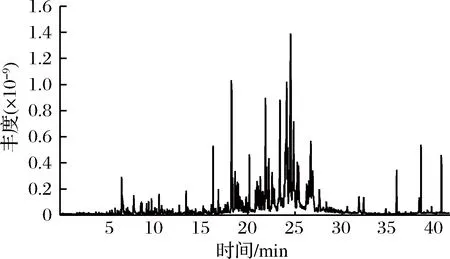
圖6 杭白菊揮發性成分總離子流圖
Fig.6 Total ion chromatogram of volatile components fromChrysanthemum moriflium Ramat
杭白菊揮發性成分復雜,各組分的相對含量差異較大,主要有烯烴類26個(26.48%)、醇類15個(23.55%)、酮類11個(5.86%)、醛類10個(2.05%)、酯類7個(5.23%)、酚類6個(3.29%)、酸類2個(0.58%)、烷烴類4個(8.09%)、芳香烴類3個(1.24%)。其中含量較高的有脫氫倍半桉樹腦(10.95%)、(2)-氧化香橙烯(10.72%)、β-欖香烯(5.90%)、α-紅沒藥醇(5.17%)、α-姜黃烯(3.43%)等。大量研究表明,β-欖香烯具有良好的抗癌活性,對多種癌細胞的增殖具有抑制作用,并且引誘細胞凋亡[27]。除此之外,β-欖香烯還具有抑制新生血管的作用,可應用于眼部疾病的治療[28]。α-紅沒藥醇具有止痛、消炎、抑菌、抗癌、抗突變、抑制遺傳損傷和去黑色素等作用[29-30]。α-姜黃烯有助于內分泌調節,而且具有抗病毒、抗腫瘤、抑癌、抑菌等生理活性[31]。另外,還測得乙酸龍腦酯(1.96%)、樟腦(1.16%)、龍腦(0.41%),據報道,乙酸龍腦酯具有鎮痛抗炎作用[32];樟腦、龍腦是揮發油抗菌作用的主要成分[33]。
3 結論
本研究以浙江桐鄉的杭白菊為研究對象,建立了HS-SPME-GC-MS/MS結合保留指數測定其中88種揮發性成分的分析方法,結果具有良好的重現性。該方法利用AMDIS的自動解卷積功能得到更加純凈的質譜圖進行譜庫檢索,再參考保留指數對組分精準定性,提高了定性分析的準確度和效率,使結果更加可靠,同時也對其他樣品的分析具有借鑒作用。
