利用CRISPR/Cas9技術敲除擬南芥轉錄因子MYB40的兩種可變剪接體
2019-01-23 00:57:08李孟湛尹紅菊李丁丁劉亞琪王鎖民
草業學報 2019年1期
關鍵詞:植物
李孟湛,尹紅菊,李丁丁,劉亞琪,王鎖民
(蘭州大學草地農業生態系統國家重點實驗室,蘭州大學農業農村部草牧業創新重點實驗室,蘭州大學草地農業科技學院,甘肅 蘭州 730020)
植物受到外界高鹽、干旱、高溫等逆境時,脅迫信號在細胞內通過一系列地傳遞,最終激發轉錄因子的表達;轉錄因子與相應的順式作用元件結合后,從而特異性地啟動應答基因的轉錄表達[1-2]。研究發現,與植物脅迫反應相關的轉錄因子主要有以下幾個家族:MYB、bZIP、WRKY、NAC類轉錄因子家族等。其中,MYB家族作為植物最大的轉錄因子家族之一,在植物抵抗非生物脅迫過程中發揮著重要功能。
研究者在模式植物擬南芥(Arabidopsisthaliana)中首次發現了受脫落酸(abscisic acid, ABA)顯著誘導的AtMYB2基因,其編碼蛋白可參與調節脫水響應基因RD22的表達[3]。近幾年先后從擬南芥、水稻(Oryzasativa)等植物中鑒定出一批參與非生物逆境響應的MYB轉錄因子,并對其作用機理進行了深入研究。如Dai等[4]在擬南芥中過量表達水稻OsMYB3R-2基因可顯著提高植株對冷、干旱和鹽脅迫的耐受力。在馬鈴薯(Solanumtuberosum)中超表達StMYB1R-1后,與干旱脅迫響應相關基因RD28、ALDH和ERD15的表達豐度大幅上調,葉片的水分散失減少,從而顯著提高了植株的抗旱性[5]。番茄(Lycopersiconesculentum) R2R3-MYB轉錄因子LeAN2受高溫誘導表達,高溫脅迫下超表達LeAN2的轉基因植株通過保持較高的鮮重、光合效率及酶活性使其耐受性增強[6]。前期研究也發現強旱生植物霸王(Zygophyllumxanthoxylum)轉錄因子ZxMYB315的表達豐度受干旱和鹽處理顯著誘導,在擬南芥中過量表達該基因顯著提高了植物對干旱和鹽脅迫的耐受力(未發表數據)。
通過ZxMYB315 CDS序列Blast比對發現,擬南芥轉錄因子MYB40與ZxMYB315的同源性最高。MYB40具有兩種不同的可變剪接體MYB40.1和MYB40.2,在擬南芥信息學分析網站eFP(http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi)上發現MYB40對鹽和干旱脅迫均有明顯響應,表明其可能在植物抗逆過程中發揮重要功能,而相關方面的研究還未見報道。因此,獲得MYB40.1和MYB40.2的敲除突變體,并以敲除突變體為材料,研究MYB40的抗逆功能具有十分重要的意義。但在ABRC(https://abrc.osu.edu/)等相關的擬南芥突變體種子庫中未找到MYB40.1及MYB40.2相應的T-DNA插入缺失突變體。鑒于此,應用CRISPR/Cas9基因編輯技術,分別獲得MYB40.1和MYB40.2的敲除突變體,為揭示MYB40這兩種可變剪接體在擬南芥響應逆境脅迫過程中的功能和分子機理奠定了基礎。
1 材料與方法
1.1 材料
本實驗所用擬南芥野生型材料為Col-0;CRISPR/Cas9中間載體AtU6-26-sgRNA-SK和終載體pCAMBIA1300-pYAO:Cas9由蘭州大學黎家實驗室提供;根癌農桿菌GV3101由蘭州大學牧草逆境生理與基因工程實驗室保存。本試驗于2017年3月至2017年11月進行。
1.2 主要試劑
植物總RNA提取試劑盒購自北京天根生化科技有限公司;引物合成及質粒提取試劑盒購自上海生物工程有限公司;膠回收試劑盒購自Omega公司;限制性內切酶BsaⅠ-HF、NheⅠ-HF、SpeⅠ-HF,堿性磷酸酶CIAP及T4 DNA連接酶購自NEB公司;PrimeScriptTMRT reagent Kit with gDNA Eraser (Perfect Real Time)、DNA marker及SYBR Green qPCR Master Mix-SYBR Advantage購自大連寶生物公司;SILWET L-77購自GE Healthcare公司;其他生化試劑為進口或國產分析純產品。
1.3 植物材料培養及處理
將健康飽滿的擬南芥Col-0種子消毒后,點種于1/2 MS固體培養基上,4 ℃春化3 d,置于培養室培養1周后收集幼苗,分別用1/2 MS液體培養基和含有150 mmol·L-1NaCl的1/2 MS液體培養基處理6 h后取樣,用于提取總RNA。
將健康飽滿的野生型擬南芥種子播種于裝有草炭土的小花盆中并放入培養室進行培養。
培養室的溫度為22~23 ℃,光照條件為120~150 μmol·m-2·s-1、16 h(晝)/8 h(夜),相對濕度50%~60%[7]。
1.4 MYB40.1及MYB40.2的表達模式分析
Real-time PCR引物設計:依據Tair網站(http://www.arabidopsis.org/)上MYB40兩種可變剪接體的cDNA序列,設計Real-time PCR引物如下(圖1,表1)。
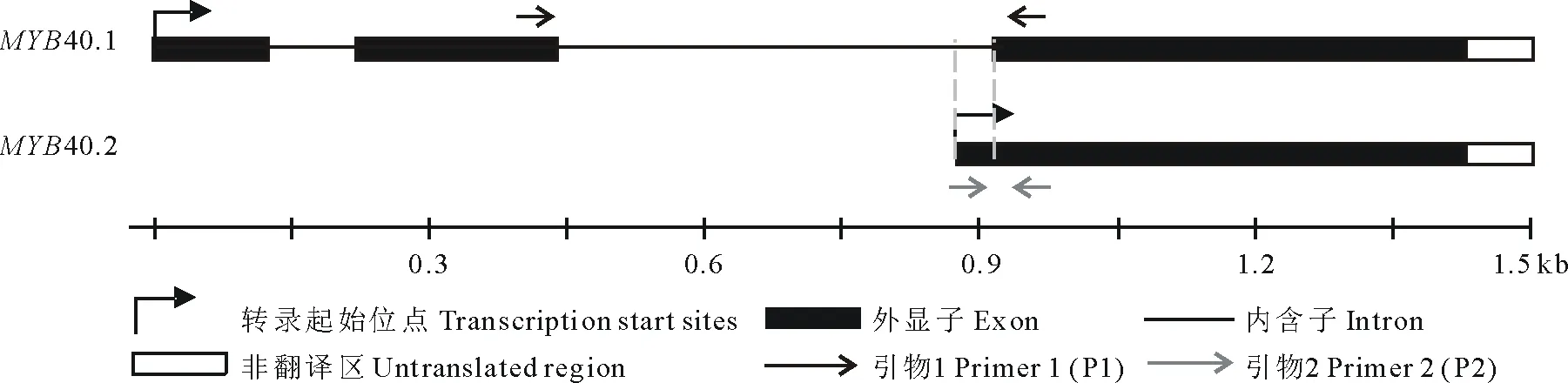
圖1 Real-time PCR引物選擇示意圖Fig.1 Selection of Real-time PCR primer
總RNA提取及cDNA獲得:取1/2 MS液體培養基及含有150 mmol·L-1NaCl的1/2 MS液體培養基處理6 h的根樣,參照植物總RNA提取試劑盒方法提取RNA并使用PrimeScriptTMRT reagent Kit with gDNA Eraser (Perfect Real Time)反轉錄獲得cDNA。
MYB40.1及MYB40.2的表達模式分析:將反轉錄獲得的cDNA稀釋5倍后作為模板進行Real-time PCR。
1.5 CRISPR/Cas9載體構建及農桿菌轉化
靶點選擇及靶點引物合成:在Tair網站上查找獲得MYB40兩種轉錄本的gDNA序列,參考Ma等[8]及Yan等[9]的靶點選擇方法,應用Optimized CRISPR Design在線設計軟件(http://crispr.mit.edu/)分別對MYB40.1、MYB40.2及MYB40進行靶點選擇。因為經BsaⅠ酶切后會留下粘性末端ATTG及AAAC,所以在正向引物的5′端添加接頭ATTG,在反向引物的5′端添加接頭AAAC。選擇MYB40.1的靶點T1.1、T1.2,MYB40.2的靶點T2.1、T2.2及MYB40.1與MYB40.2的共同靶點T(圖2),并設計相應靶點引物如下(表2)。
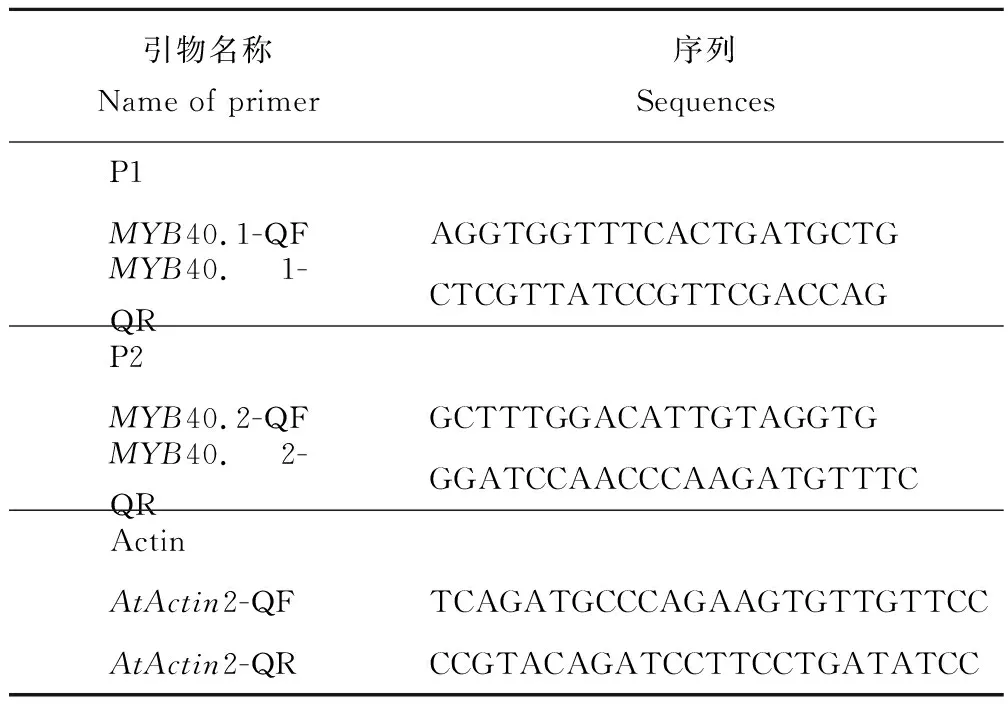
表1 Real-time PCR引物Table 1 Primers for Real-time PCR
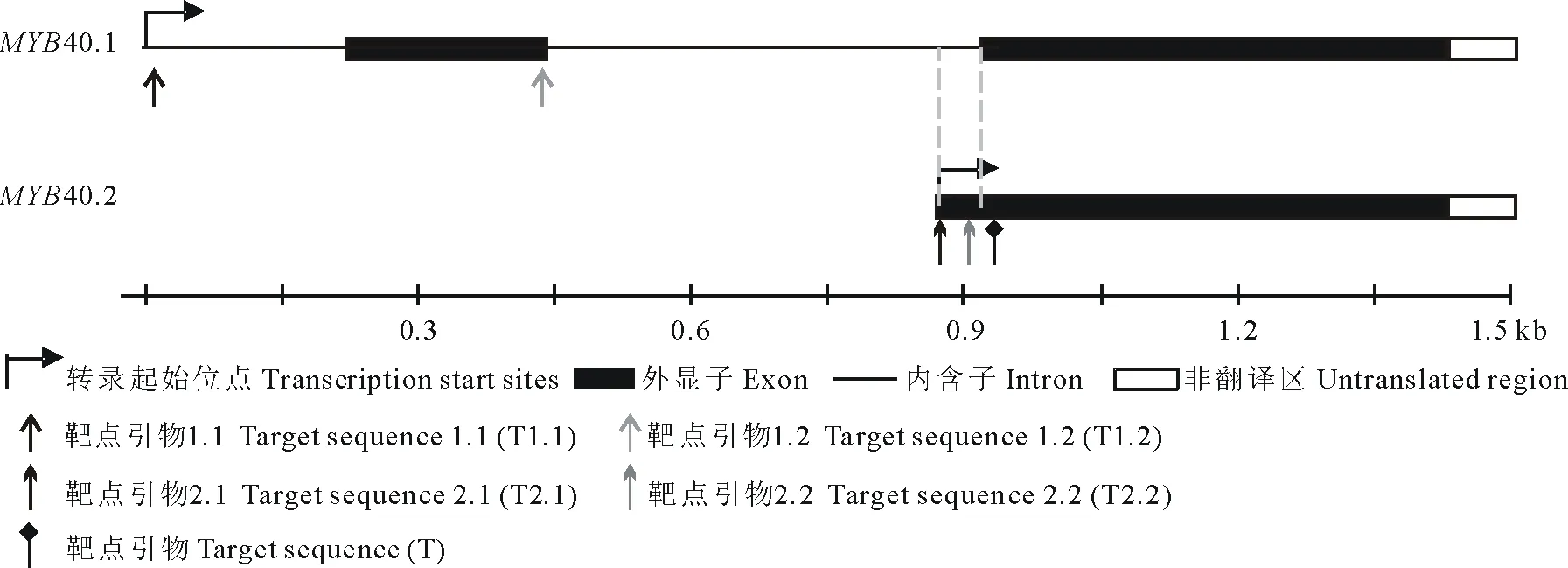
圖2 靶點選擇示意圖Fig.2 Selection of target sequences
1.5.1sgRNA cassette構建 將10 μmol·L-1的正反靶點引物各取10 μL于80 μL 0.5×TE (pH 8.0),混勻98 ℃加熱3 min后取出。取1 μg中間載體AtU6-26-sgRNA-SK于50 μL反應體系中,使用1 μLBsaⅠ-HF 37 ℃酶切4 h,凝膠電泳檢測,回收3.5 kb片段。在10 μL連接體系中,使用0.2 μL T4 DNA連接酶將40 ng酶切回收獲得的AtU6-26-sgRNA-SK片段與1 μL退火后的靶點引物在16 ℃連接過夜。
將連接產物轉化大腸桿菌DH5α感受態細胞并凃于含有50 mg·L-1氨芐的LB固體培養基上,37 ℃培養過夜,使用SK-gRNA-F (5′-CTCACTATAGGGCG AATTGG-3′)與靶點反向引物進行菌落PCR,PCR長度應為499 bp。挑選陽性克隆進行擴大培養并提取質粒送測序,將測序正確的質粒應用NheⅠ-HF和SpeⅠ-HF 37 ℃雙酶切4 h,電泳后切膠回收大小約642 bp的片段,所獲片段即為sgRNA cassette。
1.5.2雙元載體構建及農桿菌轉化 在50 μL體系中,使用1 μLSpeⅠ-HF在37 ℃酶切2 μg終載體pCAMBIA1300-pYAO:Cas9 4 h后,80 ℃失活20 min,待反應體系冷卻至室溫后,加入堿性磷酸酶CIAP 0.2 μL于37 ℃反應10 min。 電泳, 切膠回收大小約14 kb 的片段。在10 μL連接體系中,加入約40 ng酶切后的pCAMBIA1300-pYAO:Cas9及15 ng左右的sgRNA cassette,使用0.2 μL T4 DNA連接酶在16 ℃連接過夜。
將10 μL連接產物轉化大腸桿菌EscherichiacoliDH5α感受態細胞,涂于含有50 mg·L-1卡那霉素的LB固體培養基,37 ℃過夜培養。使用1300-gRNA-F (5′-CCAGTCACGACGTTGTAAAC-3′)與1300-gRNA-R (5′-CAATGAATTTCCCATCGTCGAG-3′)進行菌落PCR并進行跑膠檢測,PCR產物片段應為750 bp左右。挑選陽性克隆進行擴大培養并提取質粒。
使用凍融法將獲得的質粒轉化農桿菌GV3101,涂于含有50 mg·L-1卡那霉素及50 mg·L-1慶大霉素的LB固體培養基上,28 ℃培養48 h,用1300-gRNA-F及1300-gRNA-R引物進行菌落PCR,PCR產物片段長度約為750 bp。對于每種質粒,分別挑選陽性克隆進行擴大培養并保存于-80 ℃冰箱。
1.6 擬南芥遺傳轉化及基因編輯植株獲得
1.6.1擬南芥遺傳轉化 應用浸花法[7]對擬南芥進行轉化,4周后收集種子。
1.6.2基因編輯植株獲得 擬南芥種子消毒后,點于含有30 mg·L-1潮霉素的1/2 MS固體培養基上,4 ℃春化3 d后置于培養室生長2周,將具有潮霉素抗性的植株移植至草炭土中進行培養。
依據靶點引物的位置,在其上下游分別設計轉基因陽性植株檢測引物(表3)。SDS法[8]提取潮霉素抗性擬南芥植株葉片總DNA,使用檢測引物進行PCR檢測,PCR產物送至蘇州金維智公司進行測序分析。
2 結果與分析
2.1 MYB40.1及MYB40.2表達模式分析
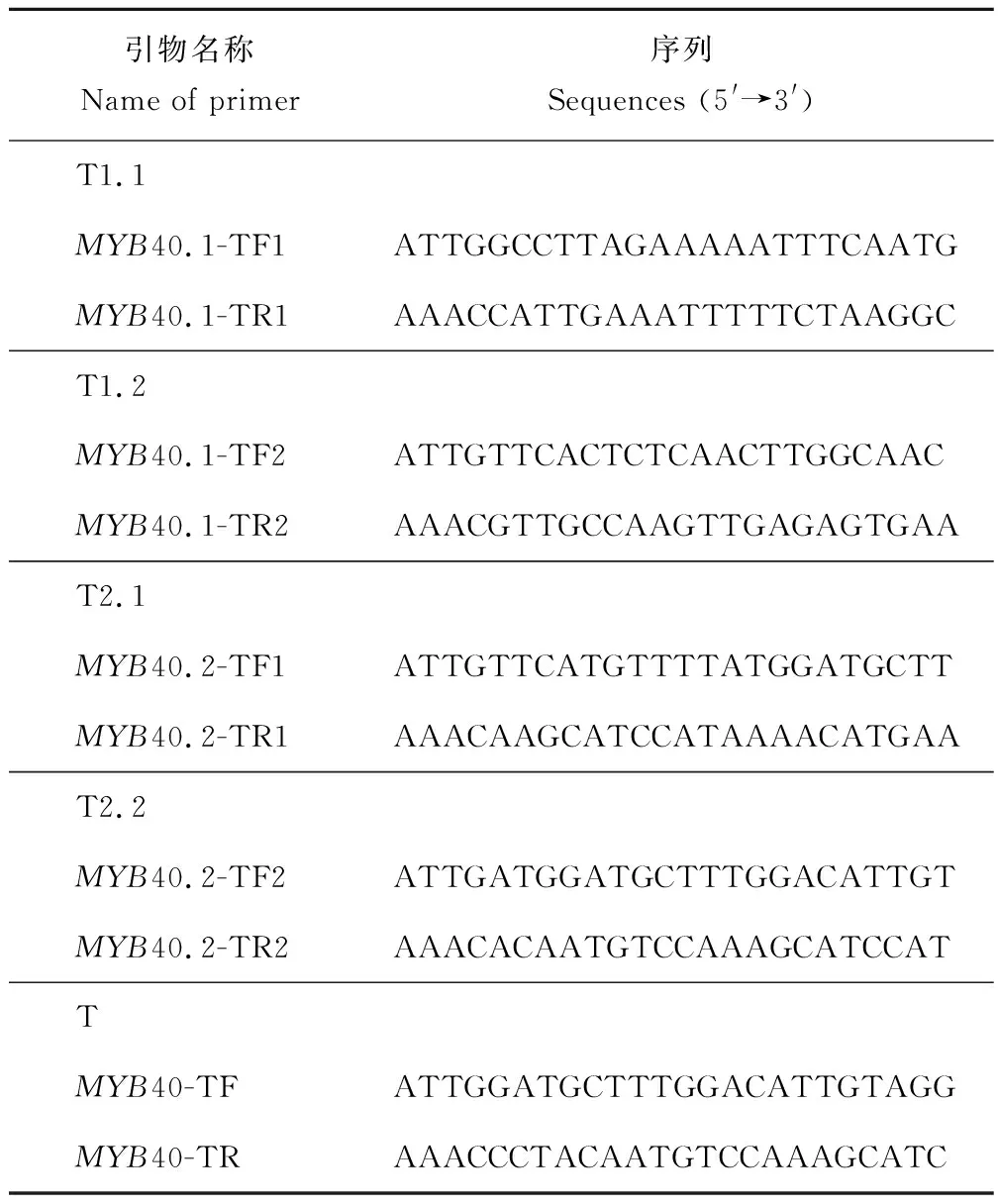
表2 MYB40不同轉錄本CRISPR/Cas9靶點引物設計Table 2 Design of CRISPR/Cas9 target primer for different transcripts of MYB40
表3 突變檢測引物設計Table 3 Design of primers for mutation detection
提取Control (1/2 MS)及150 mmol·L-1NaCl (1/2 MS+150 mmol·L-1NaCl)處理6 h擬南芥的根樣,液氮速凍后研磨,提取總RNA,使用NanoDrop 1000核酸蛋白檢測儀檢測,OD260/OD280為1.8~2.1,表明所提RNA質量較高,可用于隨后的實驗中。使用反轉錄獲得的cDNA為模板,分析鹽處理條件下MYB40.1及MYB40.2表達變化(圖3)。發現在鹽處理6 h后,MYB40.1及MYB40.2的表達均下調,表明這兩種可變剪接體在擬南芥的耐鹽過程中可能發揮了負調控的功能。
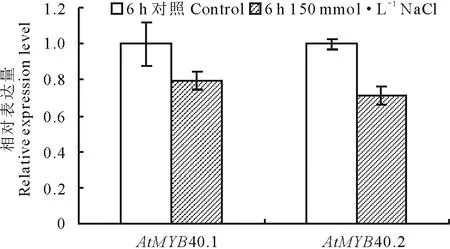
圖3 AtMYB40.1及AtMYB40.2表達模式分析Fig.3 Relative expression levels of AtMYB40.1 and AtMYB40.2
2.2 sgRNA cassette構建
通過酶切連接的方法構建sgRNA cassette。使用SK-gRNA-F與靶點反向引物進行菌落PCR,在499 bp處出現目標條帶(圖4)。對陽性克隆進行擴大培養,提取質粒后送測序,挑選測序結果合適的質粒,使用NheⅠ-HF與SpeⅠ-HF進行雙酶切,對642 bp左右的條帶進行切膠回收(圖5),所獲片段即為sgRNA cassette。
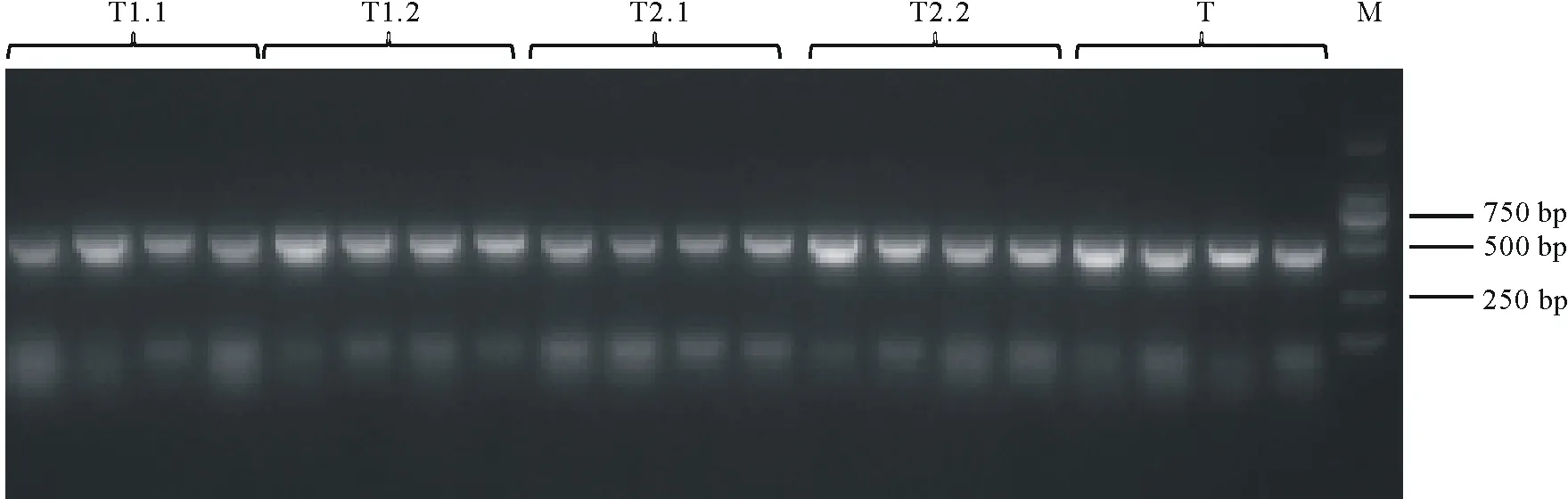
圖4 菌落PCR電泳圖Fig.4 Agarose gel electrophoresis of colony PCRM:Marker,DL 2000.下同The same below.
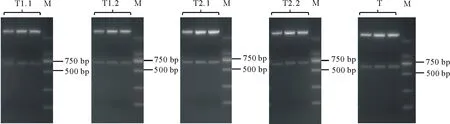
圖5 sgRNA cassetteFig.5 sgRNA cassette
2.3 雙元載體構建
通過酶切連接的方法構建雙元載體。使用1300-gRNA-F與1300-gRNA-R進行菌落PCR,在750 bp處出現目標條帶(圖6),表明MYB40兩種不同可變剪接體的CRISPR/Cas9雙元載體構建成功。
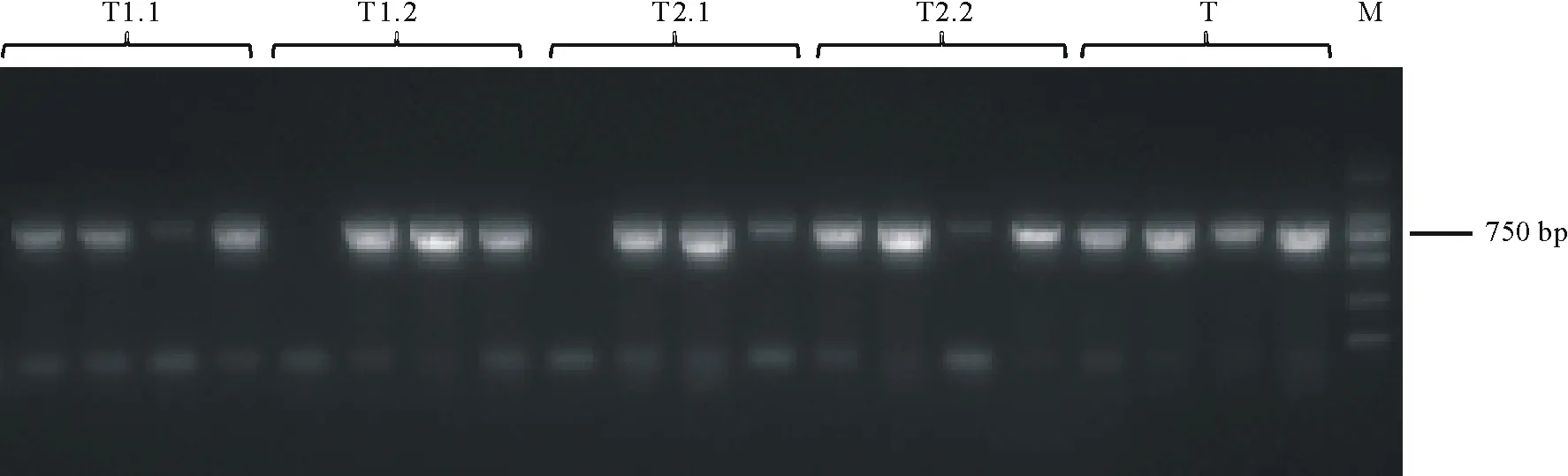
圖6 雙元載體菌落PCR電泳圖Fig.6 Agarose gel electrophoresis of binary vector colony PCR
2.4 農桿菌轉化及陽性植株鑒定
通過凍融法[6]將構建好的CRISPR/Cas9雙元載體導入根癌農桿菌GV3101中,挑取單克隆進行菌落PCR,在750 bp處出現條帶,表明雙元載體成功轉化進入根癌農桿菌中(圖7)。提取具有潮霉素抗性的擬南芥植株總DNA,并使用突變檢測引物進行檢測,PCR產物送測序,將測序結果與擬南芥野生型MYB40.1,MYB40.2的序列進行比對,序列發生變化或測序出現套峰的即為基因編輯植株(圖8)。
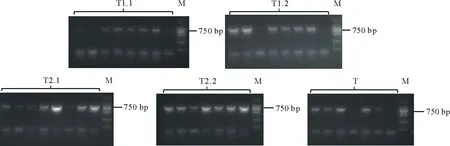
圖7 農桿菌菌檢電泳圖Fig.7 Agarose gel electrophoresis of Agrobacterium tumefaciens PCR
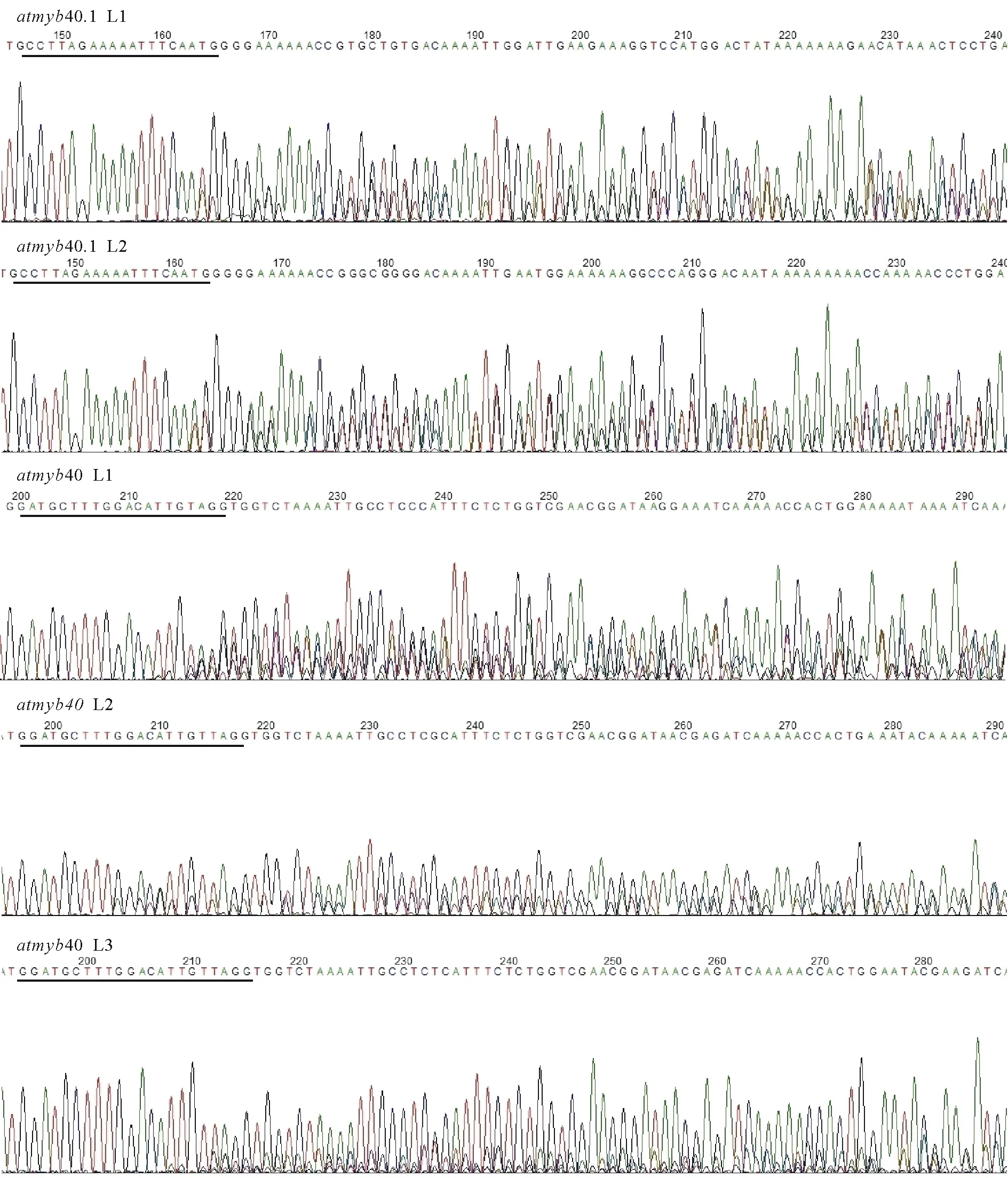
圖8 基因編輯植株測序結果Fig.8 Sequencing results of gene-edited alleles黑線標出為靶點。Black lines indicate target sequences.
3 討論
干旱、鹽堿等非生物脅迫影響了植物的正常生長發育,導致作物減產。在農業生產中,通常通過灌溉及施肥減緩不良環境條件對作物生長的不利影響[10]。這些田間管理措施雖然在短時間內提高了作物產量,但也造成了水資源浪費、次生鹽漬化及水體富營養化等危害[11-12]。因此,培育能夠保障穩定糧食供給且具有更強抗性的作物就具有十分重要的意義。植物在長期適應過程中,進化出了一系列抗逆機制并形成了相應的調控網絡。轉錄因子作為一種調控蛋白,在植物響應各種非生物脅迫的信號通路中具有十分重要的功能,其不同可變剪接體的形成在植物適應各種非生物脅迫中起到了關鍵作用[13-17]。因此,研究轉錄因子受非生物脅迫所產生的可變剪接體在抗逆過程中的功能以更好的理解植物的脅迫響應機制具有十分重要的意義。
CRISPR/Cas9技術作為一種新興的編輯技術,因其高效性及簡便性,越來越多的應用于動植物的基因功能驗證及育種中[18]。在該技術中,通過構建特定的sgRNA,引導Cas9至DNA的特定位點進行切割,形成DNA雙鏈斷裂,隨后,經非同源末端連接(non-homologous end joining,NHEJ)或同源重組(homologous recombination,HR)自我修復機制進行修復。通常情況下,NHEJ會導致隨機的插入或缺失,當插入或缺失發生在編碼區時,會導致翻譯時的移碼,無法翻譯成正確的蛋白,從而達到基因編輯的目的[19]。在使用這一技術對擬南芥的基因進行編輯的過程中,多通過農桿菌侵染花序,將包含有Cas及相關序列的載體轉入植物體內。因此,在這一過程中,T-DNA最先進入到擬南芥的胚囊細胞中[20]。然而,與其在營養組織中的活性相比,最為廣泛使用的啟動Cas表達的CaMV 35S啟動子在花序侵染的過程中活性較低,嚴重影響了編輯效率且獲得的突變多為體細胞突變(somatic mutations)。YAO參與擬南芥的胚胎發生及配子發育,主要表達于分裂能力強的組織中[21]。因此,本研究使用該基因的啟動子pYAO代替35S啟動子來啟動Cas9的表達以提高編輯效率并獲得生殖系突變(germline mutations)[9,21]。
靶位點的選擇將影響最終的編輯效率,優先選擇在內含子與外顯子的連接處、起始密碼子處及基因重要的功能結構域的NGG前設計靶點引物,使編輯后DNA轉錄獲得的pre-mRNA無法正常剪接、成熟的mRNA無法正常翻譯或翻譯獲得的蛋白不具有活性,以提高突變效率[8]。同時,為達到特異性敲除不同可變剪接體的目的,靶位點應根據兩可變剪接體序列上的差異進行選擇。因此,本研究參照Ma等[8]及Yan等[9]的方法,分別在AtMYB40.1的起始密碼子處及第2個外顯子與內含子的連接處,AtMYB40.2 CDS序列的5′端,AtMYB40.1的第3個外顯子處分別選擇編輯AtMYB40.1、AtMYB40.2及同時編輯AtMYB40.1和AtMYB40.2的靶位點。
4 結論
在本研究中,獲得3個MYB40.1和MYB40.2被同時編輯的株系及2個MYB40.1基因編輯株系,隨后將繼續對MYB40.2的突變體植株進行篩選,獲得3種不同突變體的純合植株以進行抗逆功能分析,同時,該研究也為應用CRISPR/Cas9技術對霸王等非模式植物進行基因編輯以更好的研究其抗逆機理打下基礎。
猜你喜歡
少兒科學周刊·兒童版(2021年19期)2021-12-10 14:13:40
小學閱讀指南·低年級版(2021年3期)2021-03-19 06:12:40
小天使·二年級語數英綜合(2020年8期)2020-12-23 04:57:40
小天使·一年級語數英綜合(2020年11期)2020-12-16 02:57:22
學苑創造·A版(2020年3期)2020-04-24 09:21:39
小溪流(畫刊)(2017年11期)2018-01-09 19:15:14
少兒科學周刊·兒童版(2017年5期)2017-06-29 22:24:28
少兒科學周刊·兒童版(2017年5期)2017-06-29 16:46:33
紅領巾·萌芽(2017年5期)2017-06-23 10:35:59
爆笑show(2016年7期)2017-02-09 09:36:13
