一種新的基于多重液相色譜-質譜實驗肽信號峰形相似性的校準算法
2018-12-19 06:48:02董曉睿
分析測試學報 2018年12期
崔 健,董曉睿,商 凱,陳 強,祁 鑫,崔 浩
(1.中國石油大學 勝利學院 信息技術系,山東 東營 257016;2.中國石油大學 計算機與通信工程學院,山東 青島 266580)
液相色譜-質譜(LC-MS)是發現并分析生物標志物中復雜肽信號的關鍵技術,其中對實驗譜中的肽信號進行檢測和量化至關重要。理論上相同樣本多次重復實驗得到的譜圖是一致的,即同種肽鏈應在不同譜圖的相同位置(相同LC時間與m/z值)產生相同信號。但由于實驗誤差,多次重復實驗譜圖會存在較大差異,需對譜數據進行校準,減小誤差。
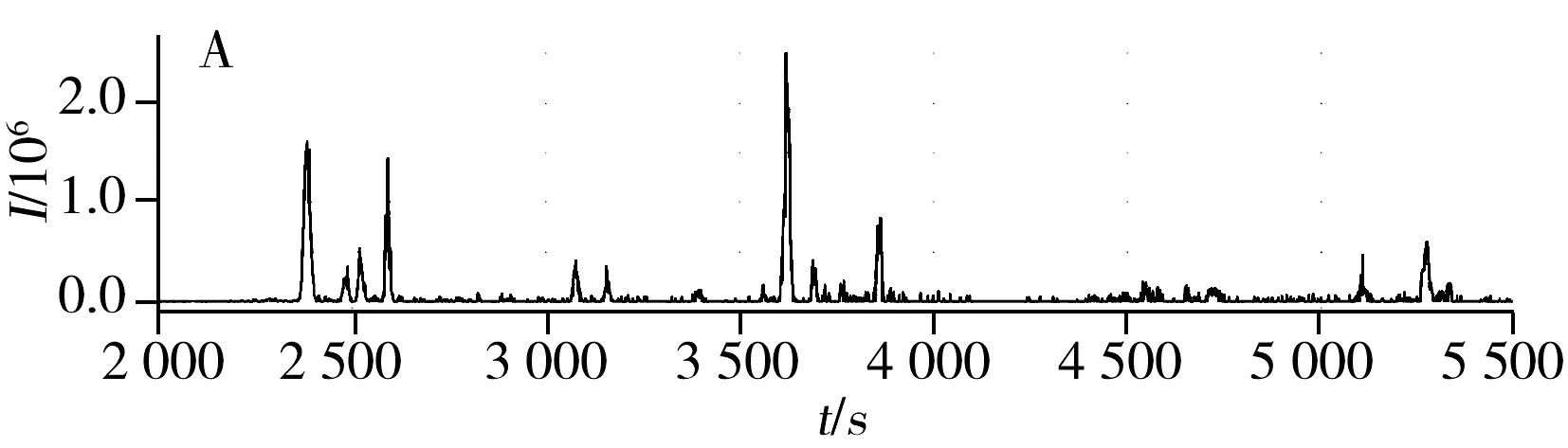
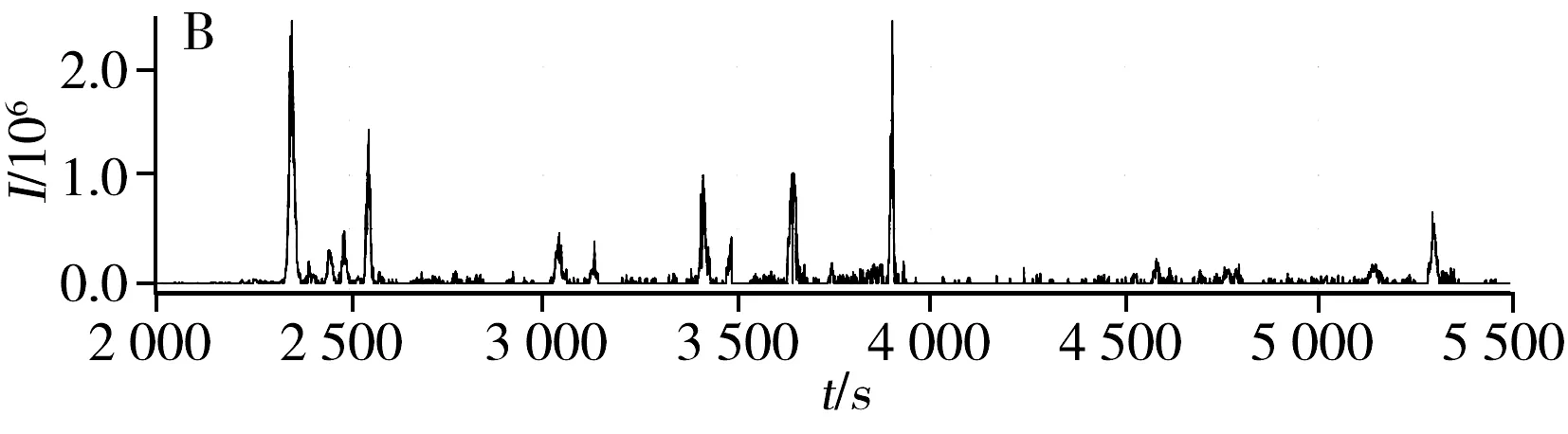
圖1 肽鏈“KVEDMMK”生成的XICsFig.1 XICs generated by “KVEDMMK”A.XICs of peptide “KVEDMMK”in data 1;B.XICs of peptide “KVEDMMK”in data 2
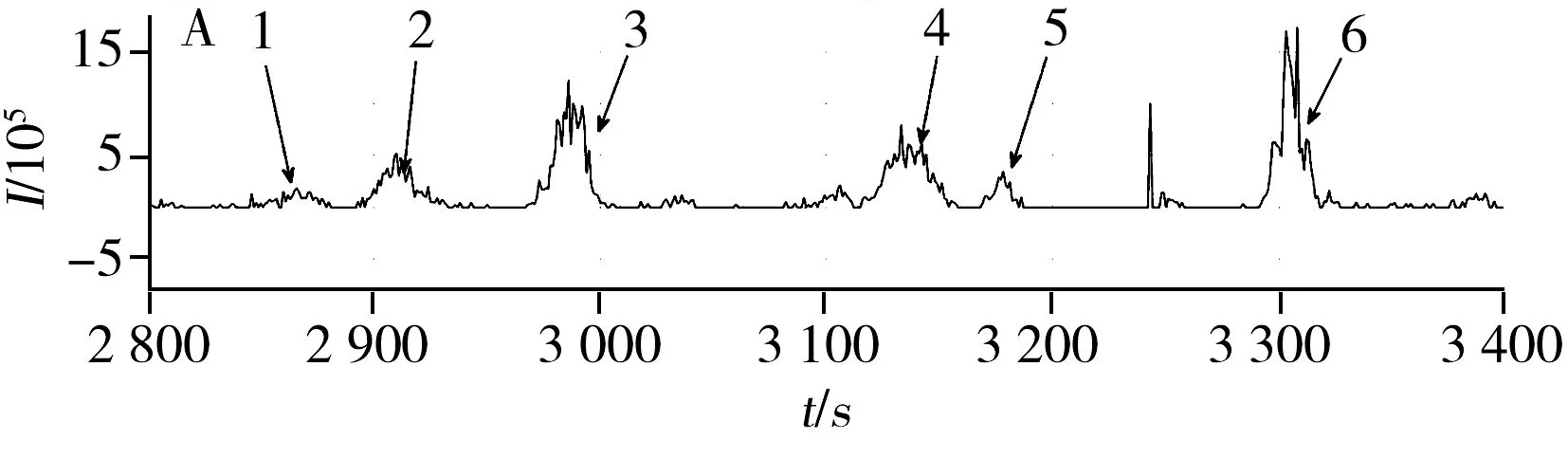

圖2 肽鏈“AGGPTTPLSPTR”的相關峰信號匹配Fig.2 Corresponding peak of “AGGPTTPLSPTR” A.LC peak between 2 800-3 400 of peptide “AGGPTTPLSPTR” in data 1;B.LC peak between 2 800-3 400 of peptide “AGGPTTPLSPTR” in data 2
根據液相色譜-二級質譜(LC-MS/MS)實驗標識的LC峰時間位置,通常使用翹曲函數(Warping function)對時間特征進行校準。目前,采用翹曲函數對LC時間軸校準,通常先計算肽鏈的m/z值,然后固定m/z值對整個時間譜圖進行匹配。但由于時間差產生的隨機性,該方法并不能完全校準。有研究者提出了基于翹曲函數的改進算法,如2002年Nielsen等[1]提出的相關優化翹曲函數算法(Correlation optimized warping,COW);2004年Eilers[2]提出的參數時間翹曲函數算法(Parametric time warping,PTW);2006年van Nederkassel等[3]提出的半參數時間翹曲函數算法(Semi-parametric time warping,STW)及Jaitly等[4]提出的液相質譜數據翹曲函數法(lcmswarp)等。Voss等[5]提出了一種將相關特征峰對和整體時間校正相結合的算法,該法關注同時校正多重實驗數據,但對數據的處理效果比OpenMS軟件[6]略差。此外,當實驗樣本比較復雜時,在一個m/z值相同的提取離子色譜圖(XICs)中會有多個LC峰出現在一個狹窄的LC時間窗口,極可能導致相應特征峰的錯誤識別。圖1顯示了肽鏈“KVEDMMK”在本文所處理的實驗數據1和2中產生的XICs,其LC峰信號充滿噪聲,主峰附近也分布很多噪聲峰,即使用OpenMS或Msinspect[7]等軟件進行多個數據集處理,也無法避免此類問題。
時間誤差的隨機性及噪聲會導致匹配結果準確性降低。以肽鏈“AGGPTTPLSPTR”為例(圖2),在數據1中檢測到多個肽鏈信號(Peak1~6),與MS/MS檢測結果對比,確認Peak3為真正的肽信號峰;數據2中由MS/MS檢測到的真實肽信號已標出。以數據2為基準匹配數據1中的真實信號,即在數據1中的6個信號中找到與數據2中真實信號相匹配的信號Peak3。而從時間間隔上顯示匹配結果為Peak2,而非Peak3。因此,除時間特征外,還需引入其他特征提高校準匹配的準確性。
目前,僅MS/MS均能識別出的肽鏈與LC-MS峰重疊的一小部分可以使用Quil[8]、Proteinquant[9]、Msinspect[10]、OpenMS[11-12]和Superhirn[13]等軟件進行重復實驗數據量化。MaxQuant軟件[14-16]可以大大提高量化范圍,是因為由MS/MS檢測的肽鏈可以至少量化1次,但在所有數據集中可以同時量化的總肽數量有限,只能是多數據MS/MS肽鏈信號的交集。這導致多次重復實驗數據量化的覆蓋率較低。
針對兩個重復LC-MS實驗數據,本文采用普通的區間檢測方法,選取多次重復實驗數據中均被MS/MS檢出的肽鏈信號作為訓練數據集。以MS/MS檢測到的肽鏈的m/z值及LC時間值為真實值(Ground truth),訓練數據集中所有肽鏈在兩個數據中均具有真實值。從訓練數據集中隨機選取部分肽鏈,基于其真實值建立統計學習模型,訓練數據集中剩余部分作為測試序列進行模型測試,以模型給出的最高分值作為匹配結果,再比對真實值,計算百分比作為檢測模型的準確性(Accuracy)。最后,僅在1個數據中被MS/MS檢測到的肽鏈通過模型匹配其在另外數據中的相關區間(無MS/MS檢測結果),提升校準后肽信號的覆蓋率。
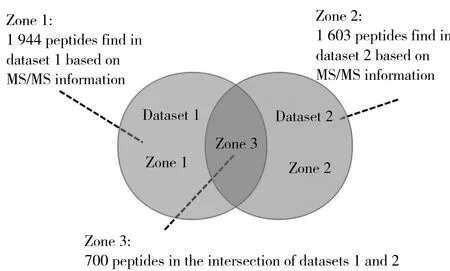
圖3 數據1和數據2的MS/MS檢測肽鏈信息文氏圖Fig.3 Venn diagram of MS/MS peptides information of data 1 and data 2
1 實驗部分
1.1 數據來源
本文處理的數據由RCMI Proteomics and Protein Biomarkers Cores at UTSA實驗室提供,為經過LTQ Orbitrap Velos儀器處理的TAGE腫瘤數據(腫瘤樣本的多次重復實驗數據),選取2組數據(數據1與數據2)進行分析。每個數據分為Level1和Level2。數據中每個數據點包括3個坐標時間值、質荷比值、強度值。數據1中Level1的有用數據(強度值不為0)為11487個,Level2的有用數據(強度值不為0)為58636個。數據2中Level1的有用數據(強度值不為0)為11446個,Level2的有用數據(強度值不為0)為59573個。數據1中被MS/MS檢測到的肽鏈為1944個,數據2中被MS/MS檢測到的肽鏈為1603個,交集為700個,并集為2847個(圖3)。
1.2 數據處理
1.2.1數據預處理如圖4所示,根據實驗1和實驗2的MS/MS信息表,生成MS/MS肽信號合集,并計算相應肽信號的質荷比(m/z值)。然后在數據1和數據2的Level 1數據中,分別計算肽鏈m/z值(前后各取20×10-6寬度)下的LC譜圖,獲取全時間段的XICs;在全時段XICs上進行區間檢測,在數據1與數據2中分別獲得的區間段均為候選信號區間。
1.2.2訓練數據集生成具備可測試的真實值是選取訓練數據集的前提。本研究以MS/MS檢測到的肽信號的m/z值與時間值為真實可靠的值。首先,選取圖3中的交集部分作為生成訓練數據集的基礎(共700個肽鏈),將預處理區間檢測后包含MS/MS時間點的肽鏈選作訓練數據集合(共599個肽鏈)。再在訓練數據集中隨機選取一半作為訓練序列,產生時間差統計學習模型以及峰形相似性模型;剩余部分作為測試序列,測試模型匹配結果的準確性(以MS/MS檢測值作為真實數據比對)。
1.2.3統計學習模型生成基于以下兩個假設建立模型:①同一肽鏈在重復實驗中產生信號區間的位置(包括m/z與時間)理論上一致,不同種肽鏈產生的信號位置有差別;②同一肽鏈重復實驗產生的信號形狀理論上一致,不同種肽鏈產生的信號形狀有差別。在訓練數據集中選取訓練肽信號k個,以圖2為例:肽鏈“AGGPTTPLSPTR”在數據1中的真實信號峰(Peak 3)與數據2中的真實信號峰為相關信號峰對,數據1中其他信號峰(除去Peak 3)與數據2中真實信號峰為非相關信號峰對,分別計算相關信號峰對的時間差與峰形相似性,以及非相關信號峰對的時間差與峰形相似性。時間差即為區間最高值的時間差值,峰形相似性為計算兩個信號的線性回歸決定系數r2的值。r2反映了兩個數列的相似程度,如數列x、y的r2值反映了數列y的變化可用數列x的變化來解釋的百分比,計算公式如下:
r2=SSreg/SStot=1-SSres/SStot
其中,SStot為總平方和,SSreg為回歸平方和,SSres為殘差平方和。r2結果在0~1之間,SStot在數據確定后始終為固定值。估計的準確性越低,則SSres越大,r2越接近0;反之,則r2越接近1,即峰形越相似r2值越接近1。
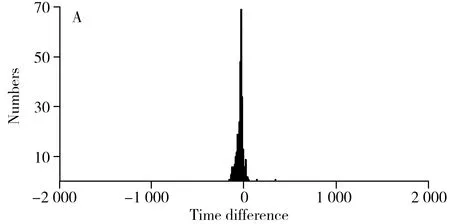
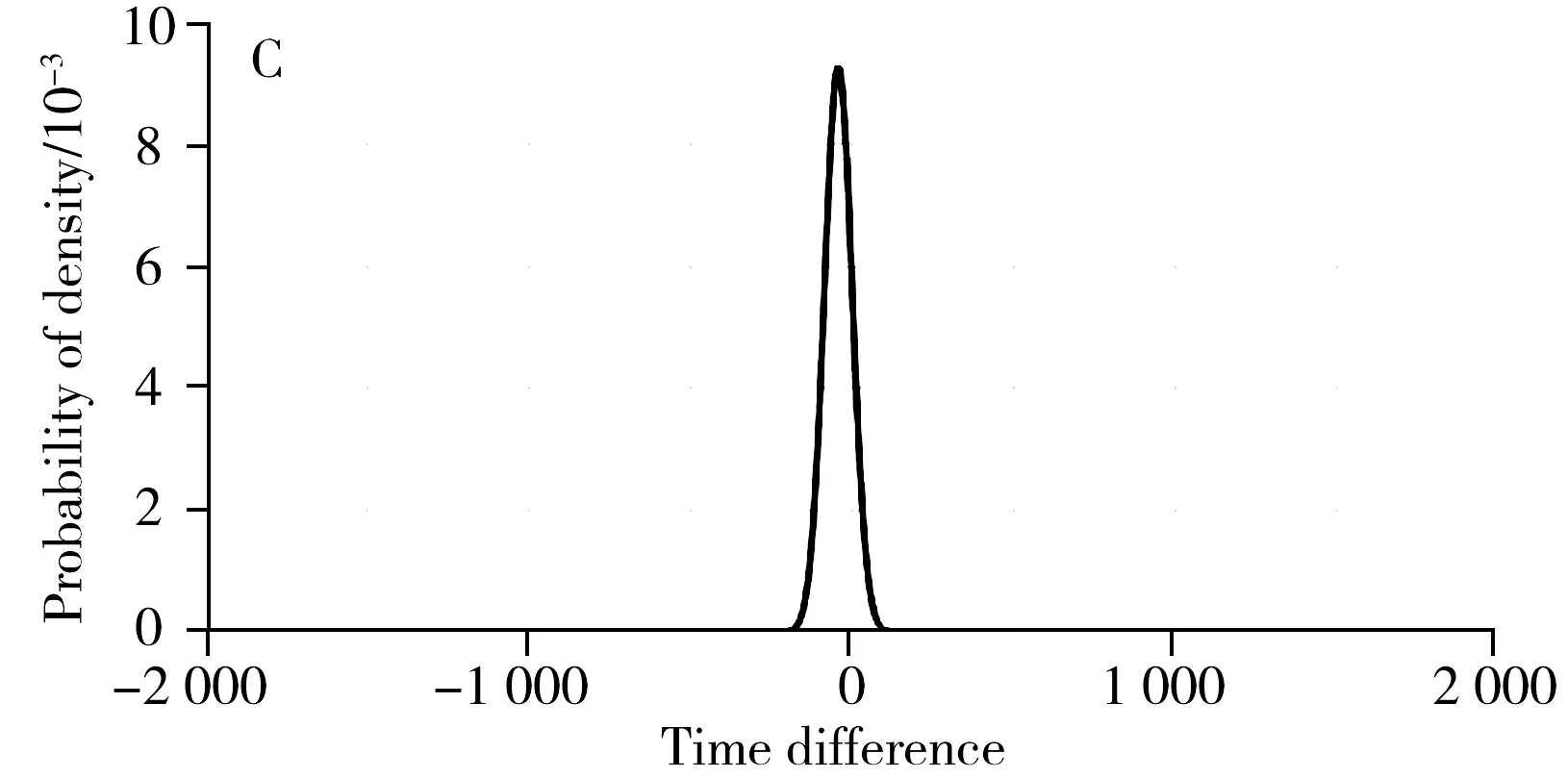
時間差特征統計特性如圖5所示。時間差直方圖基本符合正態分布,相關信號峰對的時間差相對非相關信號峰對的時間差更集中(圖5A、B)。針對時間差樣本,采用最大似然估計生成兩個正態分布模型的參數。用相關峰對的時間差樣本t估計正態分布模型f(Δt|t)的參數μ和σ:
得到相關信號峰對的時間差模型f(Δt|μ,σ2):
同理使用非相關峰對時間差樣本得到非相關信號峰對的時間差模型。圖5C、D為相關和非相關信號峰對的正態分布時間差模型。
峰形相似性特征統計特性見圖 6,相關信號峰對的相似性集中在0.7以上(圖6A),而非相關信號峰對的相似性比較分散(圖6B)。采用gamma分布進行擬合:
gamma分布具有兩個參數k和θ,采用matlab中gamfit( )函數進行gamma分布參數的極大似然估計得數值解,得到相關信號峰對的相似性模型和非相關信號峰對的相似性模型,峰形相似性模型的區分度較明顯(圖6C)。
1.2.4LC峰匹配校準基于圖3的交集數據建立時間差與峰形相似性的統計模型,并測試模型有效性后,將統計模型用于圖3中去交集部分肽鏈的校準匹配,即根據僅在1個數據中由MS/MS檢測到的肽鏈信號,匹配其在另一數據中的相關信號。最終由MS/MS檢測到的每一個肽鏈均能在數據1和2中匹配到相關信號峰對。
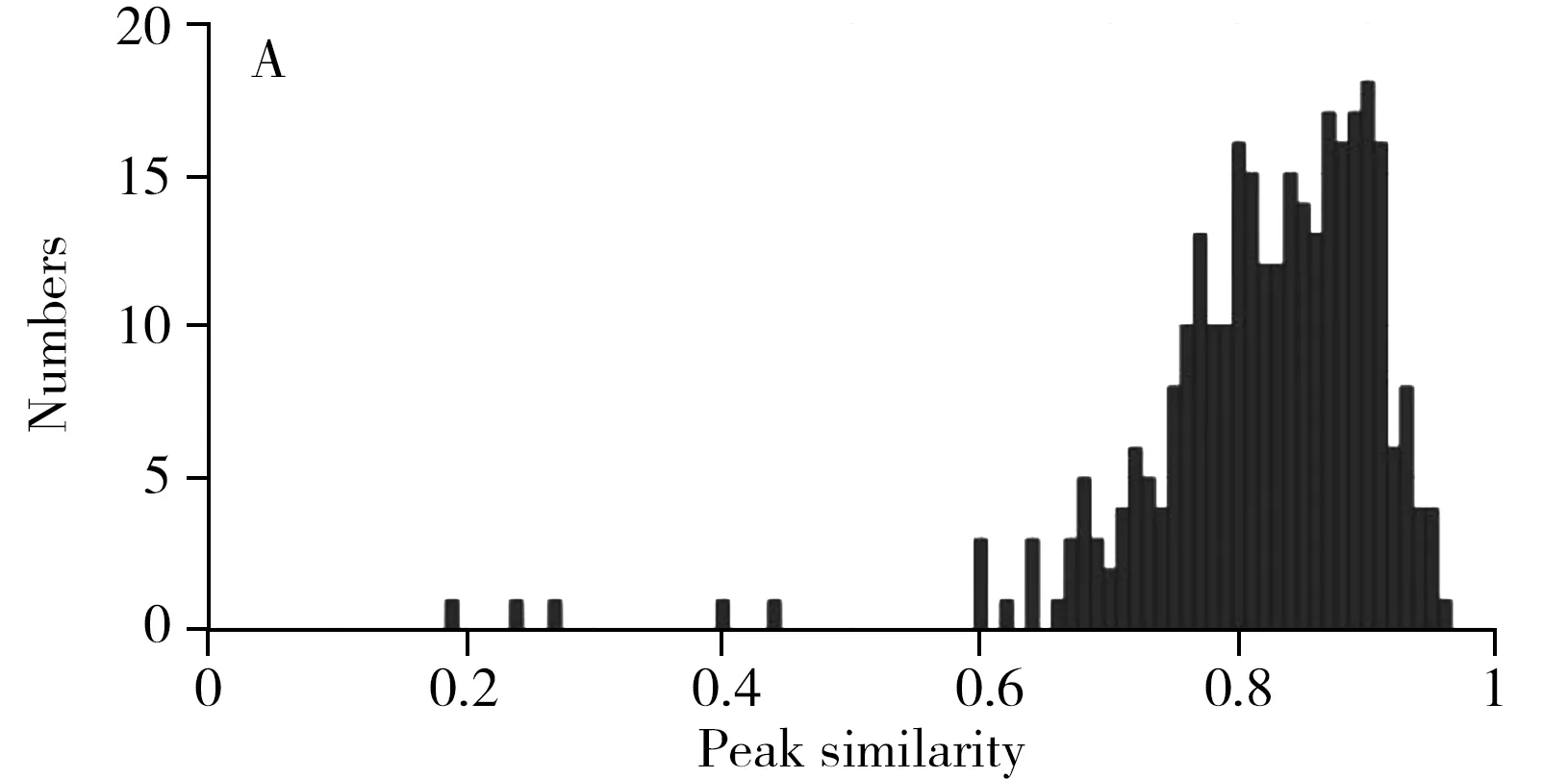
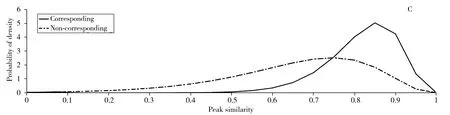
圖6 相關峰與非相關峰峰形相似性模型Fig.6 Peak shape similarity of corresponding and non-corresponding peak modelsA.histogram of corresponding peak similarity(相關峰對峰形相似性直方圖);B.histogram of non-corresponding peak similarity(非相關峰對峰形相似性直方圖);C.model of peak similarity(峰形相似性模型)
2 結果與討論
2.1 結果分析
2.1.1模型測試結果對模型進行10次測試,每次均從訓練數據集中隨機選取300個肽鏈信號作為訓練,剩余299個肽鏈信號作為測試,將模型得到的匹配結果與MS/MS檢測值(真實值)進行比對得到準確度。對時間模型和峰形模型分別進行單獨測試,然后按照峰形和時間模型不同的權重配比進行綜合測試,得到最優配比后再測試1次。
單獨使用時間模型進行10次獨立測試,準確率的平均測試值為96.3%,標準差為0.76。單獨使用峰形模型進行10次獨立測試,準確率的平均測試值為66.3%,標準差為1.79。
按照不同權重配比,綜合使用時間模型與峰形相似性模型,結果如表1所示。總體上時間模型比重越高,準確性越高,這與單獨分析時間模型結果優于峰形相似性模型的結果相符。但峰形相似性模型對整體準確性有所提升,配比為8∶2時的準確性達98.3%。

表1 模型在不同權重下測試結果Table 1 Testing result of different weights
采用8∶2的權重配比,綜合使用時間與峰形模型進行10次測試,準確率的平均值為97.8%,標準差為0.77。準確率平均值提高了1.5%,對于并集2 847個肽鏈,提高大約為42個肽鏈的校準。通過Wilcoxon rank sum test對兩種方法的結果進行顯著性檢驗:其中方法1單純使用時間方法校準,方法2采用時間峰形綜合方法校準。Wilcoxon rank sum test將觀測值和零假設的中心位置之差絕對值的秩分別按照不同的符號相加作為其檢驗統計量,檢驗成對的觀測數據之差是否來自均值為0的總體(產生數據的總體是否具有相同的均值)。本文隨機分配訓練集和測試集,共10次,方法1和方法2的結果均在具備相同訓練集和測試集的情況下得到。計算觀測數據之差,共10個樣本,假設兩種方法的結果無顯著差異,即H0:兩種方法的檢測結果無顯著差異;H1:兩種方法的檢測結果有差異;在取顯著水平為0.05的條件下,通過Wilcoxon rank sum test得到P-value為0.001,h值為1,即在5%的顯著水平下拒絕H0,表明方法2相比方法1的結果有明顯改進。
2.1.2數據并集的校準匹配根據圖3,數據1與數據2并集共4 247個肽鏈,區域1中有1 944個(數據1中待校準匹配的個數),區域2中有1 603個(數據2中待校準匹配的個數),并集共3 547個肽鏈。通過算法匹配,最后得到匹配區間的肽鏈共3 226對,校準匹配的覆蓋率達91.0%。
2.2 討 論
以上研究存在以下問題:
①區間檢測準確性需提高。本文的區間檢測是以基礎峰值位置檢測到高強度峰區域內背景噪聲標準偏差的3倍作為閾值,高于閾值的信號被納入區間,長度超過連續6個點的信號被認為是候選LC峰區間。但數據1和數據2的MS/MS交集共700個肽鏈,僅檢測到599個包含MS/MS時間點區間的肽鏈,檢測覆蓋率為85%。
②數學模型區分度需提高。通過驗證,時間差模型區分度好,但在噪聲較多的XICs中,仍受大量干擾信號影響。峰形相似性模型的引入雖有所改善,但區分度比時間差模型差。本文僅用線性回歸決定系數(r2)描述兩個信號相似性的值,模型的準確性有待提高。
③雙模型的混合應用。本文對兩個模型的綜合使用采取簡單的設置權值后相加的方法,今后將探索建立一個數學模型對兩個特征進行統一。
3 結 論
本文通過采用統計學習的方法,利用多次重復的液相色譜-質譜實驗的譜圖中肽信號的時間差與峰形相似性兩個特征,選取訓練數據集建立統計模型,并對模型有效性進行驗證,完成了對譜圖的校準,并實現了多個肽信號對的匹配,準確性達98%以上,覆蓋率達91.0%,為基于多次重復LC-MS實驗數據的肽鏈量化提供了有意義的數據支撐。
致 謝:Michelle Zhang及RCMI Proteomics and Protein Biomarkers Cores at UTSA實驗室為本研究提供了生物實驗數據,并為論文寫作與修改提供巨大幫助。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
電子制作(2018年11期)2018-08-04 03:25:42
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
