高效液相色譜法分析2,6-二氨基-3,5-二硝基吡啶-1-氧化物的純度
2018-11-12 03:37:52張蒙蒙王友兵周杰文
火炸藥學(xué)報(bào) 2018年5期
張蒙蒙,張 林,王友兵,周杰文,李 媛
(1.西安近代化學(xué)研究所,陜西 西安 710065; 2.空軍駐西北地區(qū)軍事代表室,陜西 西安 710043)
引 言
隨著人們對(duì)彈藥安全性能的重視,各國(guó)在高能低易損彈藥研究方面投入了大量人力和物力。高能量低感度炸藥是低易損彈藥研究的物質(zhì)基礎(chǔ)和關(guān)鍵。2,6-二氨基-3,5-二硝基吡啶-1-氧化物(ANPyO)是一種新型高能低感炸藥,自合成報(bào)道以來(lái)得到了廣泛關(guān)注,在混合炸藥、共晶共混材料方面具有潛在的應(yīng)用價(jià)值[1-4]。
目前,ANPyO的研究主要集中在合成工藝[5-6]、精制方法[8-9]、金屬配合物[10]、混晶[4]及熱分解性能[11]等基礎(chǔ)研究方面,應(yīng)用研究也逐步開(kāi)展[12-13]。含能材料性能與化合物純度關(guān)系密切,ANPyO的純度分析對(duì)其應(yīng)用性能具有重要影響。周心龍等[9]提出了以甲醇為流動(dòng)相的液相色譜分析法,由于ANPyO難以溶解于甲醇,該方法存在以下不足:一方面樣品在用甲醇配置溶液時(shí)不能全部溶解于溶劑,測(cè)試結(jié)果與實(shí)際情況存在誤差;另一方面色譜譜圖峰型較低,影響測(cè)試結(jié)果的精度。張蒙蒙等[8]曾報(bào)道了一種利用核磁氫譜儀測(cè)試ANPyO純度的方法,但該法默認(rèn)樣品中僅含ANPyO與雜質(zhì)ANPy,且核磁氫譜法測(cè)試化合物純度并未得到廣泛認(rèn)可。
液相色譜法測(cè)試含能化合物純度的方法已經(jīng)獲得廣泛應(yīng)用[14-16], 由于ANPyO難溶于常見(jiàn)有機(jī)溶劑,常規(guī)液相色譜法測(cè)量其純度存在困難。而純度分析是ANPyO深入研究的基礎(chǔ),隨著相關(guān)應(yīng)用研究的推進(jìn),獲得準(zhǔn)確高效ANPyO純度的分析方法顯得更加迫切。本研究采用可微溶ANPyO的二甲基亞砜(DMSO)作為樣品溶劑,利用自行建立的高效液相分析方法開(kāi)展了ANPyO純度分析,建立了純度分析方法,可為其質(zhì)量控制提供參考。
1 實(shí) 驗(yàn)
1.1 試劑與儀器
ANPyO、ANPy,均為自制;二甲基亞砜、乙腈、甲醇,均為色譜純,美國(guó)Fisher公司;實(shí)驗(yàn)用水為二次純水,并經(jīng)0.45μm濾膜過(guò)濾。
LC-20AT高效液相色譜儀,日本島津株式會(huì)社;CQ-300AD超聲儀,上海躍進(jìn)醫(yī)療器械有限公司;BSA224S分析天平,賽多利斯(北京)有限公司。
1.2 色譜條件
反相色譜柱:InertSustain C18柱(5μm, 4.6mm×250mm);流動(dòng)相:甲醇與水的體積比為40∶60;流速1mL/min;檢測(cè)波長(zhǎng)315nm;柱溫為30℃;進(jìn)樣量為10μL。
1.3 實(shí)驗(yàn)過(guò)程
分別稱取ANPyO標(biāo)樣0.0025、0.0050、0.0100、0.0150、0.0200、0.0250g(精確至0.0002g)置于50mL容量瓶中,用二甲基亞砜溶解后稀釋至標(biāo)線,震蕩均勻,獲得質(zhì)量濃度為20、50、100、200、300、400、500mg/L的ANPyO溶液,作為標(biāo)準(zhǔn)測(cè)試溶液。在上述色譜條件下進(jìn)行測(cè)定,每個(gè)質(zhì)量濃度的標(biāo)準(zhǔn)溶液平行進(jìn)樣3次,記錄色譜峰高,分別取3次平行結(jié)果的算術(shù)平均值作為最終結(jié)果。
2 結(jié)果與討論
2.1 色譜條件的選擇
2.1.1 檢測(cè)波長(zhǎng)的選擇
為獲得較佳的液相色譜檢測(cè)波長(zhǎng),用HPLC自帶二極管陣列,對(duì)ANPyO及ANPy樣品吸光度進(jìn)行測(cè)量。ANPyO及ANPy的分子結(jié)構(gòu)如圖1所示;其對(duì)應(yīng)的紫外吸收?qǐng)D譜如圖2所示。

圖1 ANPyO及ANPy的分子結(jié)構(gòu)Fig.1 Molecular structures of ANPyO and ANPy
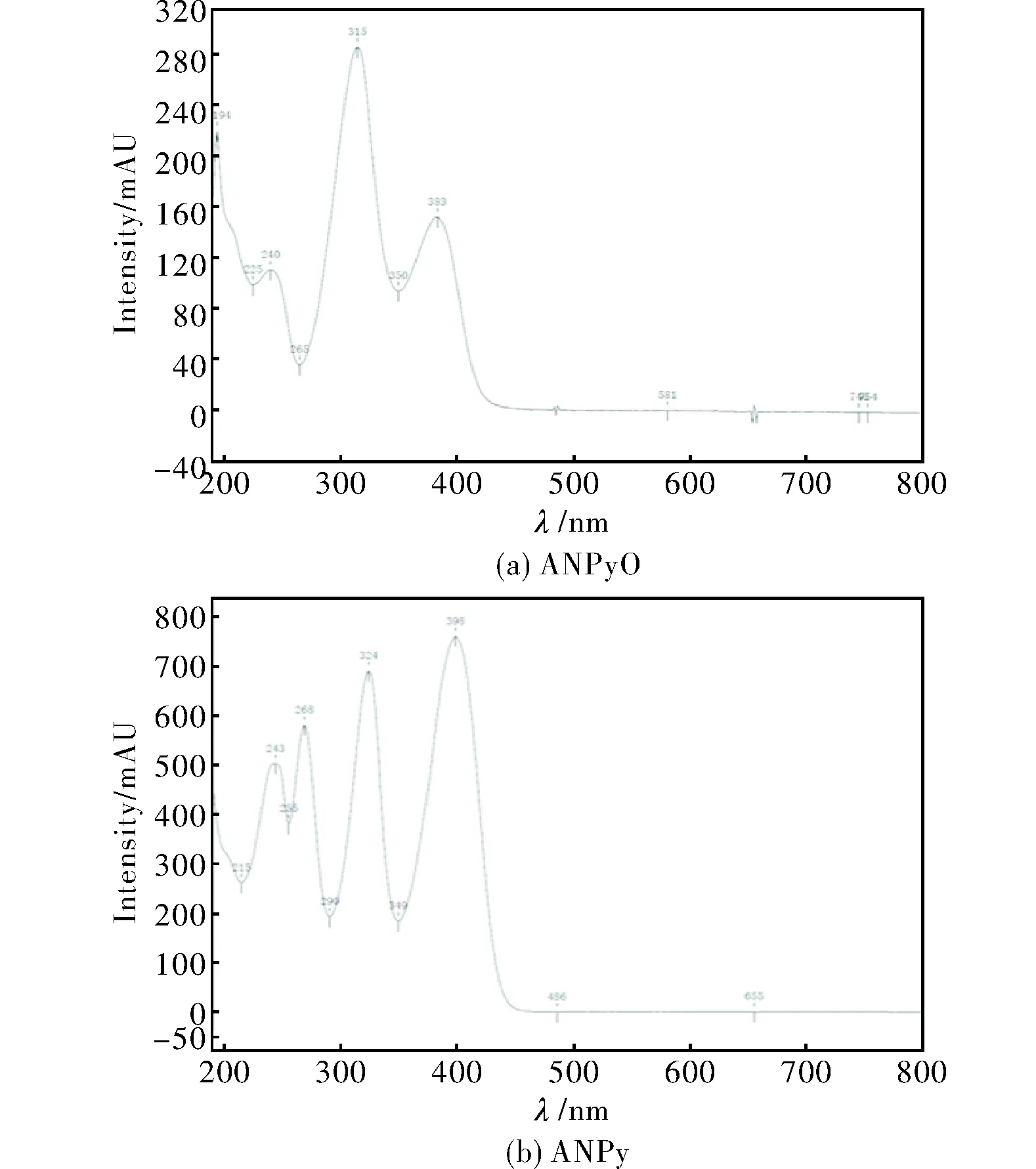
圖2 ANPyO及ANPy的紫外光譜Fig.2 Ultraviolet scanning spectra of ANPyO and ANPy
由圖2可知,ANPyO最大吸收波長(zhǎng)為315nm;ANPy有多個(gè)吸收峰,最大吸收波長(zhǎng)為398nm。由于檢測(cè)的主要成分是ANPyO,同時(shí)在315nm處ANPy有可觀的吸收峰值,選擇315nm為檢測(cè)波長(zhǎng)。
2.1.2 流動(dòng)相的選擇
反相液相色譜常用溶劑為乙腈、甲醇、水。本研究分別用乙腈/水、甲醇/水兩組混合溶劑為流動(dòng)相,開(kāi)展了流動(dòng)相篩選研究。首先選擇乙腈/水為流動(dòng)相,并通過(guò)調(diào)節(jié)其比例開(kāi)展ANPyO樣品的液相色譜分析,如圖3(a)所示。
由圖3(a)可知,當(dāng)乙腈體積分?jǐn)?shù)低至40%時(shí),ANPyO主峰存在拖尾,且各組分仍不能徹底分開(kāi)。在此色譜條件下對(duì)甲醇/水體系進(jìn)行嘗試,結(jié)果如圖3(b)所示,ANPyO主峰雖有拖尾,各單組分可分離。分析認(rèn)為,ANPyO分子內(nèi)相鄰的—NO2、—NH2、—N—O形成分子內(nèi)氫鍵,相比于ANPy,其分子中所有的氨基氫原子參與形成了分子內(nèi)氫鍵,其與色譜柱的作用力小于ANPy,因此在流動(dòng)相的作用下ANPyO先于ANPy流出。兩者由于結(jié)構(gòu)相似度高,在分離過(guò)程中往往存在流出峰重疊的情況,因此流動(dòng)相的選擇顯得尤為重要。據(jù)文獻(xiàn)[17]報(bào)道,甲醇極性參數(shù)為6.6,乙腈為6.2,在反相色譜柱中,極性越強(qiáng),對(duì)溶質(zhì)的洗脫能力越弱,出峰越晚,極性略強(qiáng)的甲醇中溶質(zhì)出峰稍晚,在出峰延遲的過(guò)程中ANPyO與ANPy實(shí)現(xiàn)了分離。因此,選擇甲醇/水為ANPyO液相色譜分析的流動(dòng)相進(jìn)一步優(yōu)化。
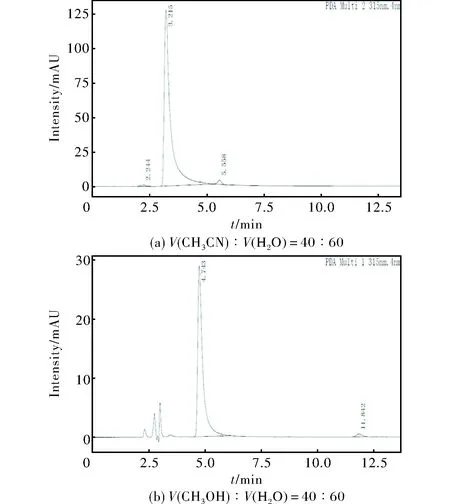
圖3 不同流動(dòng)相的ANPyO色譜譜圖Fig.3 HPLC spectra of ANPyO with different mobile phases
2.1.3 樣品溶劑的選擇
由于樣品ANPyO在乙腈和甲醇中的溶解度較小,一方面不便于樣品溶劑的制備,另一方面導(dǎo)致出峰信號(hào)過(guò)低,對(duì)檢測(cè)結(jié)果造成不利影響。本研究采用強(qiáng)極性溶劑DMSO對(duì)ANPyO進(jìn)行溶解,將制備得到的ANPyO溶液滴入體積分?jǐn)?shù)為40%的甲醇水溶液中,靜置24h未發(fā)現(xiàn)固體析出。同時(shí),為了降低色譜純DMSO對(duì)ANPyO液相色譜分析結(jié)果的影響,在以上獲得的液相條件下進(jìn)行了空白實(shí)驗(yàn),所得譜圖見(jiàn)圖4。結(jié)果表明,在2.5~3.5min處有明顯的出峰。在后期純度分析時(shí),通過(guò)色譜條件調(diào)整,避免待檢測(cè)物與DMSO出峰重合,結(jié)果計(jì)算時(shí)需剔除DMSO的影響。
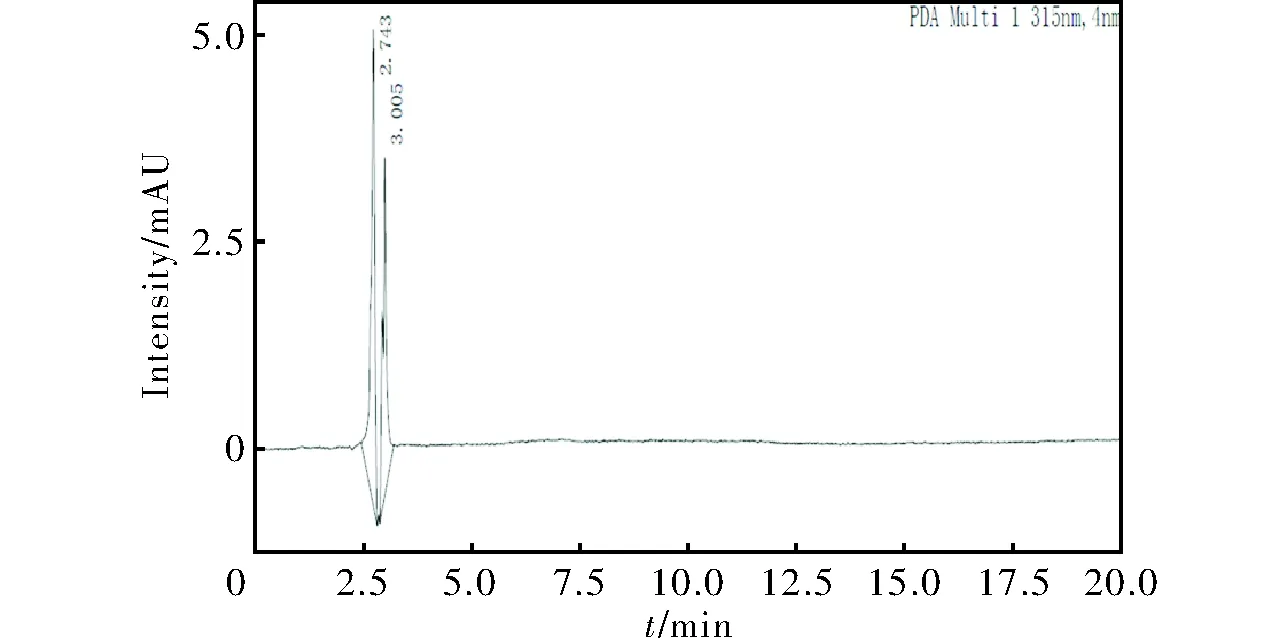
圖4 DMSO的高效液相色譜圖Fig.4 HPLC spectrum of DMSO
2.1.4 流動(dòng)相體積比的選擇
為保證樣品中各組分具有較好的分離度,探索了甲醇與水體積比分別為80∶20、50∶50、40∶60、20∶80時(shí)的色譜分離情況,結(jié)果如圖5所示。由圖5可知,當(dāng)甲醇體積分?jǐn)?shù)低于50%時(shí),ANPyO樣品中各組分可分開(kāi)。其中,甲醇與水體積比為80∶20時(shí),主峰與雜質(zhì)可分開(kāi),但流出時(shí)間長(zhǎng)達(dá)15min,而體積比為50∶50時(shí),主峰與雜質(zhì)峰有部分重合。對(duì)比分析,當(dāng)甲醇與水體積比為40∶60時(shí),流出時(shí)間適宜,各組分分離較好,可作為較優(yōu)流動(dòng)相比例條件。


圖5 不同流動(dòng)相體積比的ANPyO液相色譜圖Fig.5 HPLC spectra of ANPyO with different volume ratio of CH3OH/H2O in mobile phases
2.1.5 柱溫度及進(jìn)樣量的選擇
當(dāng)色譜柱柱溫過(guò)高時(shí),會(huì)導(dǎo)致色譜柱使用壽命縮短,而溫度低于環(huán)境溫度則不利于色譜柱的穩(wěn)定,因此,本研究選擇30℃作為液相色譜柱保溫溫度。由于樣品采用極性溶劑DMSO溶解,為降低其對(duì)設(shè)備及測(cè)試結(jié)果的影響,同時(shí)獲得合適的出峰信號(hào),進(jìn)樣量選擇為10μL。
2.1.6 流速的選擇
以甲醇與水(體積比為40∶60)為流動(dòng)相、柱溫為30℃時(shí),考察了流速分別為0.5、1.0、1.5mL/min對(duì)色譜譜圖的影響。結(jié)果表明,隨著流速的增加,樣品的保留時(shí)間明顯縮短,峰面積減小。流速過(guò)大會(huì)使樣品與雜質(zhì)的保留時(shí)間較短且過(guò)于接近,從而造成基線分離效果不佳,另外與溶劑DMSO出峰存在部分重合,影響結(jié)果的準(zhǔn)確性。同時(shí)流速過(guò)大會(huì)使色譜儀壓力過(guò)大從而影響色譜儀的穩(wěn)定性,造成分析方法的精密度下降。較低的流速使樣品與雜質(zhì)分離得更徹底,但卻導(dǎo)致檢測(cè)時(shí)間過(guò)長(zhǎng)從而降低實(shí)驗(yàn)效率。綜合考慮這兩方面的因素,確定流速為1mL/min。
2.2 線性關(guān)系
采用DMSO沉降法對(duì)ANPyO精制兩次,核磁氫譜法及以上條件歸一化測(cè)得ANPyO純度大于99.9%。將其制備為不同質(zhì)量濃度的ANPyO的DMSO溶液,作為標(biāo)準(zhǔn)測(cè)試溶液。在以上較優(yōu)條件下,每個(gè)質(zhì)量濃度的樣品平行進(jìn)樣3次,記錄色譜峰高,并繪制質(zhì)量濃度與液相色譜峰面積的關(guān)系圖,見(jiàn)圖6。
由圖6可見(jiàn),在20~500mg/L范圍內(nèi),ANPyO的峰面積與質(zhì)量濃度呈良好的線性關(guān)系,回歸方程為Y=60584X+2×106,相關(guān)系數(shù)R2為0.997。
2.3 準(zhǔn)確度及精密度
采用外標(biāo)法,對(duì)同一樣品進(jìn)行5次平行試驗(yàn)驗(yàn)證,得到的數(shù)據(jù)如表 1所示,測(cè)試結(jié)果的偏差小于0.5%,相對(duì)標(biāo)準(zhǔn)偏差≤0.24%,表明該方法測(cè)試準(zhǔn)確性和精密度較高。

圖6 ANPyO質(zhì)量濃度與色譜峰面積的標(biāo)準(zhǔn)曲線Fig.6 Standard curve of ANPyO mass concentration to corresponding chromatographic peak area
2.4 合成過(guò)程樣品檢測(cè)
ANPyO的合成參照文獻(xiàn)[18]進(jìn)行,當(dāng)反應(yīng)溫度達(dá)到80℃后,分別取2、4、6h時(shí)刻反應(yīng)體系中固體,經(jīng)過(guò)濾洗滌干燥后分別采用核磁法[8]和外標(biāo)法對(duì)樣品純度進(jìn)行分析,結(jié)果如表2所示。
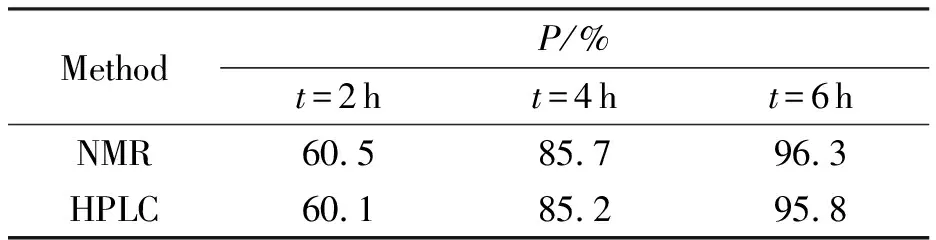
表2 核磁法和液相色譜法獲得的樣品純度
由表2可知,核磁法和液相色譜法測(cè)得的樣品純度相當(dāng),核磁法所得純度均略高于液相色譜法,主要是核磁法默認(rèn)樣品中只含有ANPyO和ANPy,而研究建立的液相色譜法測(cè)試結(jié)果包含了其他雜質(zhì),所得結(jié)果更符合實(shí)際情況。
3 結(jié) 論
(1)以二甲基亞砜為溶劑,在C18色譜柱中,甲醇與水的體積比為40∶60,液相流速1mL/min,檢測(cè)波長(zhǎng)315nm,柱溫為30℃,進(jìn)樣量為10μL的優(yōu)化條件下,建立了分析主成分2,6-二氨基-3,5-二硝基吡啶-1氧化(ANPyO)和主要雜質(zhì)2,6-二氨基-3,5-二硝基吡啶(ANPy)的新的高效液相色譜分析方法。
(2)該方法具有線性范圍較寬、重現(xiàn)性好、結(jié)果準(zhǔn)確的優(yōu)點(diǎn),可用于ANPyO的純度分析。
