氣—質聯用法同時檢測高脂食品中14種光引發劑
2018-10-10 10:47:36徐文泱
食品與機械 2018年8期
徐文泱 陳 華
(湖南省食品質量監督檢驗研究院,湖南 長沙 410017)
光引發劑是紫外光印刷油墨中非常重要的組成部分,它受紫外光照射后,吸收光的能量,分裂成2個活性自由基,引發光敏樹脂和活性稀釋劑發生連鎖聚合,使膠黏劑交聯固化,作為油墨固化的催化劑被廣泛用于紫外光油墨的印刷包裝上[1]。常見的小分子光引發劑主要分為水性均裂碎片型光引發劑和水性氫轉移型光引發劑。前者有水性安息香衍生物類(如安息香、安息香雙甲醚、安息香乙醚、安息香異丙醚、安息香丁醚)、苯乙酮衍生物(如α,α-二乙氧基苯乙酮、α-羥烷基苯酮、α-胺烷基苯酮、二苯基乙酮、α,α-二甲氧基-α-苯基苯乙酮)以及烷基苯酮類,水性氫轉移型光引發劑的代表化學結構為蒽醌、硫雜蒽酮以及二苯甲酮,其中水性二苯甲酮類又可按其在二苯甲酮母核引入的基團分為陽離子、陰離子和非離子類型[2]。光引發劑因其合成較為簡單,品種多樣,價格低廉而備受市場的青睞。
但由于光引發劑是溶脂性小分子物質,極易從包裝材料遷移到食品中,造成食品污染[3]。已有報道證實食品中也能檢測出二苯甲酮、異丙基硫雜蒽酮等光引發劑[4]。試驗[5-6]表明光引發劑苯甲酮可增加罹患腫瘤、尿道下裂的風險,引發多種過敏性皮膚病。因此,食品中多種光引發劑檢測方法的建立可以為此類污染物的風險監測提供技術支持,提高食品質量安全監管能力,健全預警應急機制。
目前關于光引發劑的檢測方法以食品包裝材料方向居多[7-9]。劉艷等[10]建立了塑料及紙塑類包裝材料中9種光引發劑的氣相色譜質譜法。在食品中檢測光引發劑的方法研究相對較少,主要集中在乳制品及飲料中光引發劑的檢測[11-13]上。前處理方法主要有固相萃取、固相微萃取技術和QuEChERS方法等,使用的儀器多為氣相色譜—質譜聯用儀和液相色譜串聯質譜儀。張耀海等[11]研究了8種光引發劑在橙汁、蘋果汁、桃汁、菠蘿汁和涼茶5種食品中的GC/MS-MS檢測方法,結果表明,方法的回收率偏差較大,最低為60.4%,最高達到99.1%,結果的相對偏差最高接近16%。另有文獻[12]采用GC-MS法對乳制品中的異丙基硫雜蒽酮檢測方法進行了報道,檢出限在0.007 mg/kg左右。
由于光引發劑多為脂溶性小分子物質,其向高脂高蛋白的食品中遷移概率更高。本研究擬選取14種有代表性的酮、醚、酯類光引發劑,拓展了研究食品種類,針對肉類、乳制品、糕點等高油富含蛋白質的食品,著力于建立一種靈敏度高、選擇性好,回收率和精密度等技術參數均能達到方法學指標要求的高通量GC-MS檢測方法,以同時檢測出高脂食品中的14種光引發劑,為研究此類光引發劑的遷移規律提供依據。
1 材料與方法
1.1 材料與試劑
2-氯噻噸酮、苯甲酰甲酸甲酯、2-苯甲酰苯甲酸甲酯、2-甲基二苯甲酮、4-氯-二苯甲酮、四乙基米氏酮、2-羥基-甲基苯丙酮、2,4-二乙基硫雜蒽-9-酮、3甲基-二苯甲酮、米氏酮、4-甲基二苯甲酮、安息香二甲醚、二苯甲酮、對二甲氨基苯甲酸異辛酯:99%,德國Dr Ehrenstorfer公司;
丙酮、正己烷、乙酸乙酯、石油醚、甲醇:色譜純,上海安譜實驗科技股份有限公司;
HLB小柱:60 mg,3 mL,上海安譜實驗科技股份有限公司;
PSA試劑管:2 mL(150 mg MgSO4,50 mg PSA,50 mg GCB,50 mg C18),上海安譜實驗科技股份有限公司;
試驗用食品:市售。
1.2 儀器與設備
氣相色譜—質譜聯用儀:Agilent 7890A-5975C MSD型,美國安捷倫科技公司;
高速冷凍離心機:Neofuge 1600R型,力康生物醫療科技控股有限公司;
分析天平:SPS401F型,奧豪斯儀器(上海)有限公司;
渦混振蕩儀:CM-1000型,東京理化器械株式會社。
1.3 方法
1.3.1 色譜條件 色譜柱:Agilent 19091S-433(30 m×250 μm×0.25 μm);進樣口溫度:250 ℃;柱流速:1 mL/min。進樣量:1 μL;程序升溫條件:初始溫度為60 ℃保持1 min,以20 ℃/min升至180 ℃保持3 min,以5 ℃/min升至280 ℃保持5 min。
1.3.2 質譜條件 EI 源。離子源溫度為230 ℃,四極桿溫度150 ℃,分別采用SCAN模式和SIM模式,在整個確認試驗中,14種光引發劑的色/質譜圖信息見表1。針對檢測的光引發劑品種多,保留時間相對集中的特點,在選擇掃描離子時考慮以下幾個原則:① 盡量選擇豐度高的碎片離子;② 選擇的監測離子扣除背景后,其信噪比應盡量大于3。
當試樣待測液保留時間和目標化合物一致(±0.5%),其質譜碎片離子質荷比吻合,豐度比與標準品的偏差符合表2,則可對試樣待測液中的光引發劑進行定性確認。
1.3.3 標準溶液的配制 準確稱量14種標樣各0.1 g(精確至0.000 1 g)于10 mL容量瓶中,稀釋溶劑為丙酮,各光引發劑母液濃度為10 g/L。分別移取0.1 mL母液于100 mL容量瓶中,以丙酮定容至刻度,此為標準儲備液,各光引發劑的濃度均為10 μg/mL。系列工作溶液現配現用。
1.3.4 樣品前處理 將樣品勻質后,準確稱取2~5 g(精確至0.000 1 g),以20 mL丙酮—正己烷(體積比2∶8)提取,渦混振蕩15 min后,于4 000 r/min離心5 min,取上清液加入到活化后的HLB小柱,以10 mL丙酮—正己烷進行洗脫,收集洗脫液,濃縮至近干以丙酮定容到2 mL,移取1 mL到PSA凈化管中,離心后上機。
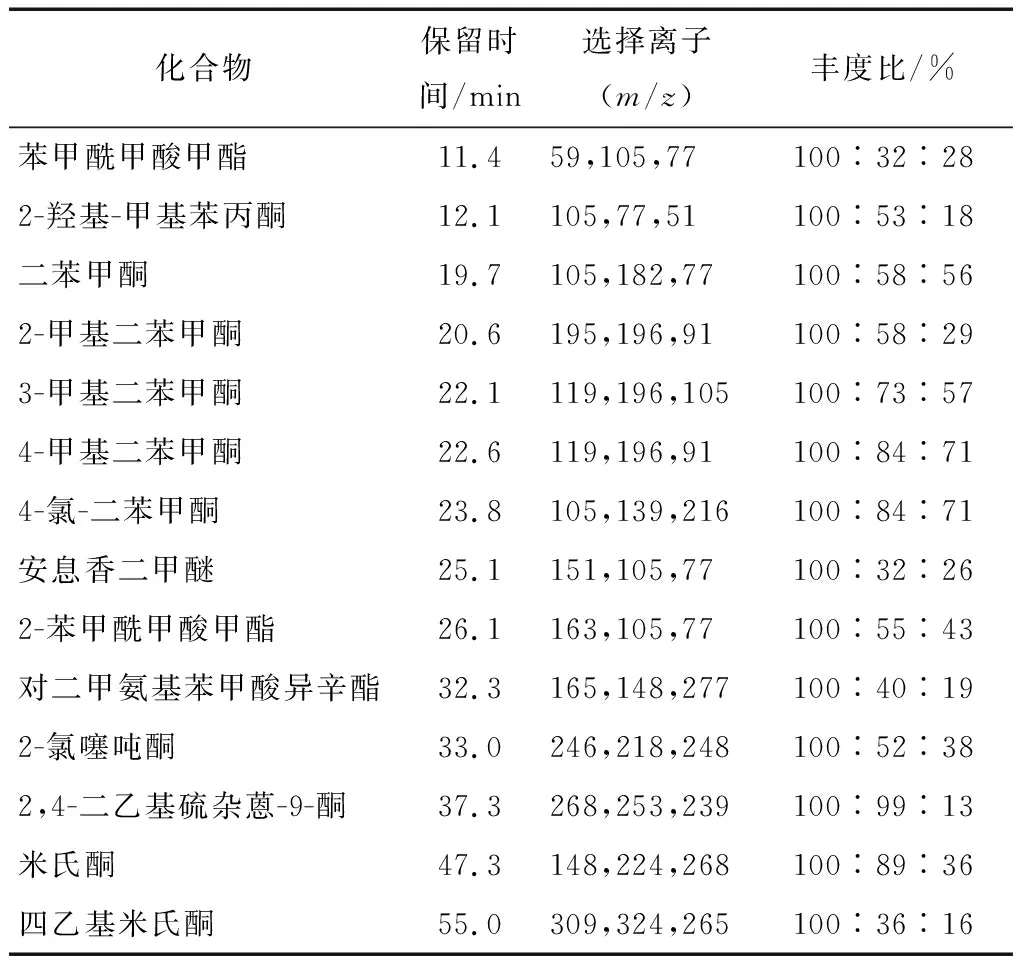
表1 14種光引發劑的保留時間和質譜中的選擇離子及豐度比Table 1 Retention time and selected ion of 14 photoinitiators
表2氣相色譜—質譜定性確證相對離子豐度最大容許誤差
Table 2 The maximum allowable error of relative ion abundance by gas chromatography-mass spectrometry %

相對豐度GC-MS相對離子豐度最大允許誤差(50,∞)±10(20,50]±15(10,20]±20[0,10]±50
2 結果與討論
2.1 色譜條件的優化
試驗考察了2種不同的色譜程序升溫條件:
(1) 60 ℃保持1 min,以20 ℃/min到150 ℃保持1 min,再以2 ℃/min 升到270 ℃保持8 min。
(2) 60 ℃保持 1 min,以 20 ℃/min 到 240 ℃ 保持0 min,再以2 ℃/min 到 270 ℃保持25 min。
從圖1、2可以看到,在第(1)種程序升溫條件下14種光引發劑僅出現了9個峰,且苯甲酰甲酸甲酯和2-羥基-甲基苯丙酮的2峰分離度差,在第(2)種程序升溫條件下14種光引發劑均能出峰,但峰與峰過于緊密,影響定量分析。因此將第(2)種程序升溫條件進行改進。將出峰較多的前段升溫程序放緩,即當60 ℃保持1 min后,以10 ℃/min 到120 ℃保持2 min,再以5 ℃/min 到240 ℃保持5 min,以2 ℃/min升溫至270 ℃保持15 min。由圖3可以看到,14種光引發劑均能檢測出,并且峰形良好,分離度較佳。
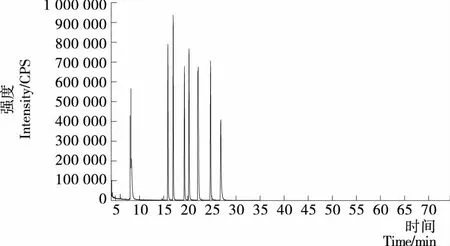
圖1 程序升溫條件(1)的總離子流圖
圖1 GC-MS chromatogram of the mixture of 14 photoinitiators with 1stcolumn temperature programme
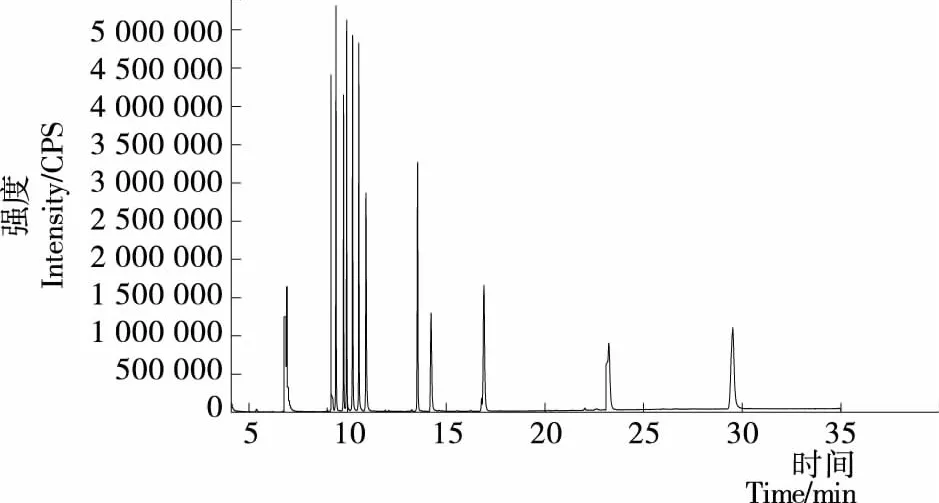
圖2 程序升溫條件(2)的總離子流圖
圖2 GC-MS chromatogram of the mixture of 14 photoinitiators with 2ndcolumn temperature programme
14種光引發劑均為脂溶性化合物,而高脂高蛋白食品種類繁多,基質相對復雜。而采用氣/液相質譜聯用儀定量分析復雜基質中化合物含量時應考慮基質效應。將空白樣品經過前處理的提取凈化以及濃縮至近干后,分別加入相同體積不同濃度的標準溶液,振蕩離心后即得相應質量濃度的基質標準工作溶液。按式(1)對基質效應的程度進行評估。當ME≥20%時,需考慮基質效應對結果準確度帶來的影響。由表3可知,ME均小于20%,在定量分析中無需考慮基質效應。可以看到本試驗前處理條件下測得的光引發劑檢出限低于文獻[14]報道,對目標物的痕量檢測具備一定優勢。
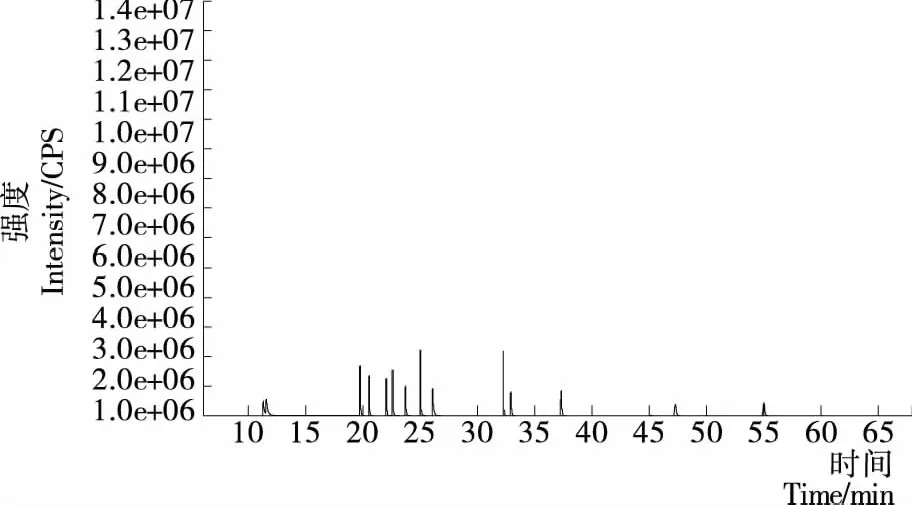
圖3 程序升溫條件(3)的總離子流圖
圖3 GC-MS chromatogram of the mixture of 14 photoinitiators with 3rdcolumn temperature programme
(1)
式中:
ME——基質效應,%;
M1——基質匹配校準曲線斜率;
M2——純溶劑標準曲線斜率。
2.2 提取條件的確認
選取的14種光引發劑酮類、酯類結構居多,并有少量硫醚類物質。根據文獻[15-17]及其化學性質,選取甲醇、乙酸乙酯、丙酮以及乙腈為參考提取溶劑。在配制標準溶液過程中發現,2-氯噻噸酮不溶于甲醇和乙腈,改選用極性稍弱的丙酮和乙酸乙酯。由于乙酸乙酯的出峰時間早于苯甲酰甲酸甲酯的出峰時間,導致了它可能被乙酸乙酯的溶劑峰覆蓋,且硫醚類物質在乙酸乙酯溶液中不穩定。因此,初步選擇丙酮為試驗的提取溶劑。由于高脂食品易發生乳化現象,在用有機溶劑萃取前先加入少量氯化鈉以達到破乳的目的。已有研究[14]表明,非極性溶劑和中等極性溶劑的結合使用有利于提高目標提取物的穩定性,并且有效地促進目標物和雜質的分離。因此,選取體積比1∶1的丙酮—正己烷、丙酮—石油醚和丙酮—二氯甲烷作為提取溶劑進行回收率試驗。提取和凈化操作按1.3.4進行,圖4為不同的提取溶劑與回收率關系示意圖。
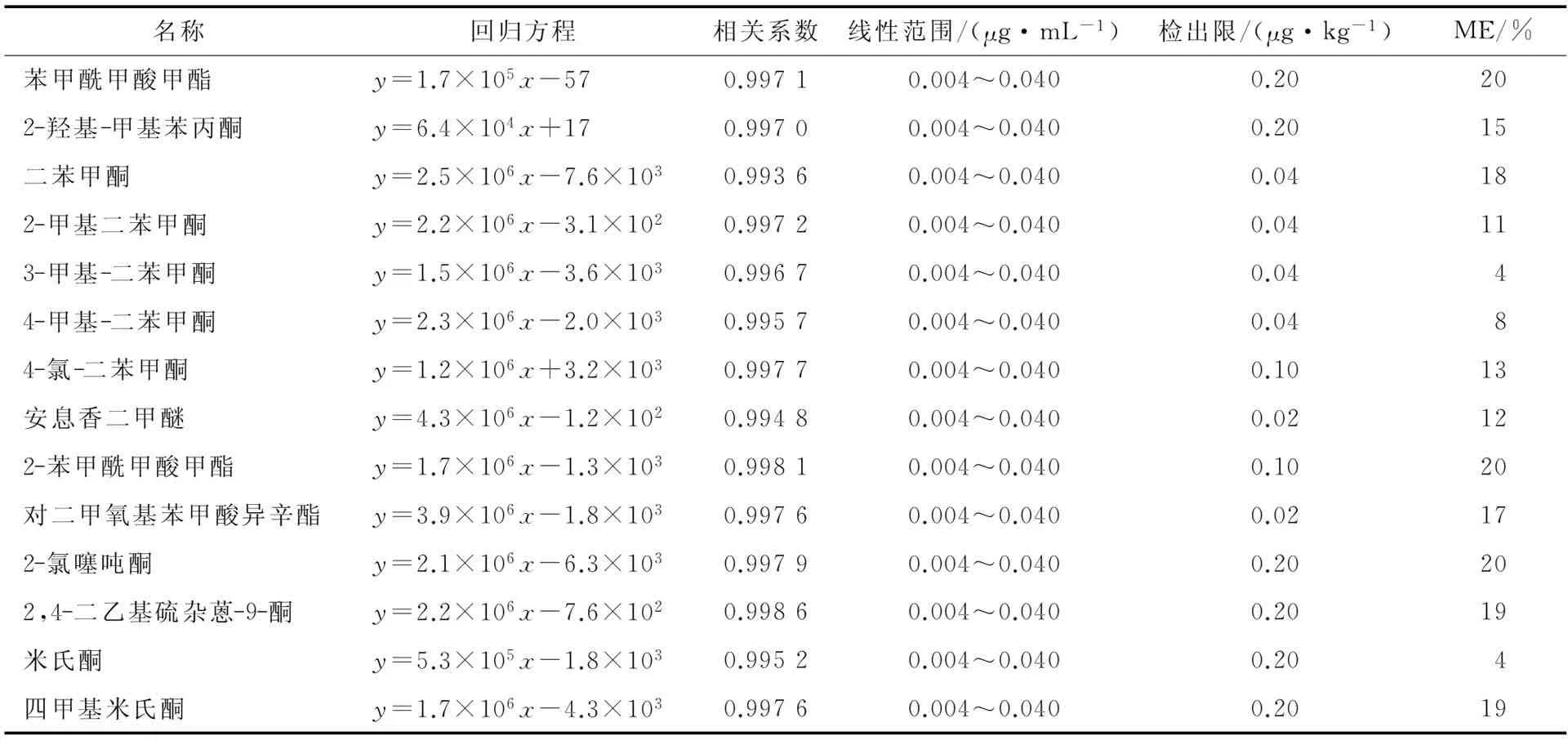
表3 14種光引發劑的回歸方程、相關系數、線性范圍及檢出限Table 3 Linear equation, correlation coefficients, linearity range and LOD of 14 photoinitiators
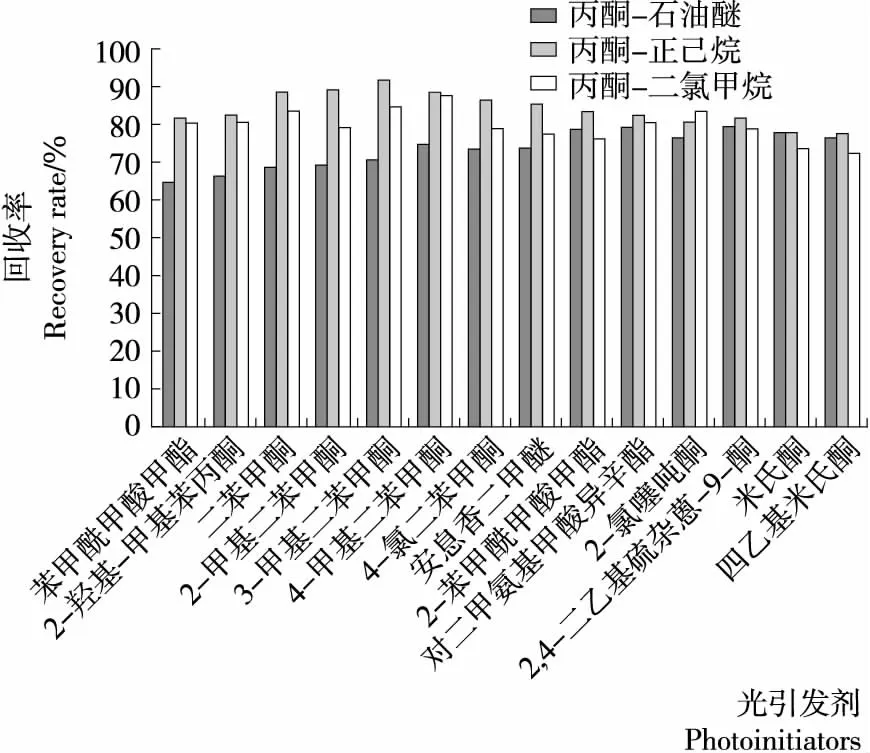
圖4 不同的提取溶劑對回收率的影響Figure 4 Effects of different solvents on the recoveries
由圖4可知,丙酮—正己烷對14種光引發劑的平均回收率最高。進一步建立正交試驗法考察萃取條件。分別選取提取溶劑的體積比、萃取溫度、提取時間作為萃取條件的3個影響因素,設計了三因素三水平正交試驗。試驗設計見表4。試驗結果和極差分析見表5。
結果表明,丙酮的比例減少有利于回收率的提高,可能是提取效率較高,在提取了目標物的同時雜質也較易溶在此溶劑中。由表5可知,最佳提取條件為丙酮和正己烷體積比2∶8,萃取溫度20 ℃,提取時間15 min。按該條件進行3次平行驗證實驗,光引發劑的平均回收率為93.1%,優于其他試驗條件所得的回收率,因此確認提取條件為以丙酮—正己烷(體積比2∶8)為提取溶劑,20 ℃下提取15 min即可最大效率地提取出所有目標化合物。
2.3 凈化條件的優化
2.3.1 吸附劑的選擇 QuEChERS方法是分散固相萃取技術的衍生和發展,其核心成分是N-丙基乙二胺吸附劑。目前,在食品及包裝材料中光引發劑檢測的報道中用到此類凈化方法的較多[11,18]。由于本試驗中食品基質較為復雜,采用單一的QuEChERS方法進行凈化可能無法取得較為理想的凈化效果。因此,嘗試采用HLB小柱和QuEChERS技術相結合的模式進行凈化,以達到去除基質干擾,降低檢出限的目的。由圖5可以看出,單采用QuEChERS方法凈化在加標濃度較低時的回收率大部分均低于采用兩者相結合的方法進行凈化的。HLB柱中的吸附劑是親水親脂平衡聚合物,它的作用基團是苯基、乙烯基和吡咯烷酮基,應用機理是通過非極性的相互作用來保留目標物。在其凈化的基礎上用PSA進行下一步凈化以去除碳水化合物、脂肪酸、有機酸、酚類和色素,可有利于提高凈化效果,去除基質的干擾,也能提高方法靈敏度,達到痕量分析的目的。圖5的回收率也可顯示將這兩種凈化吸附劑結合起來使用效果最優。

表4 正交試驗因素水平表Table 4 Factors and levels of orthogonal experiment
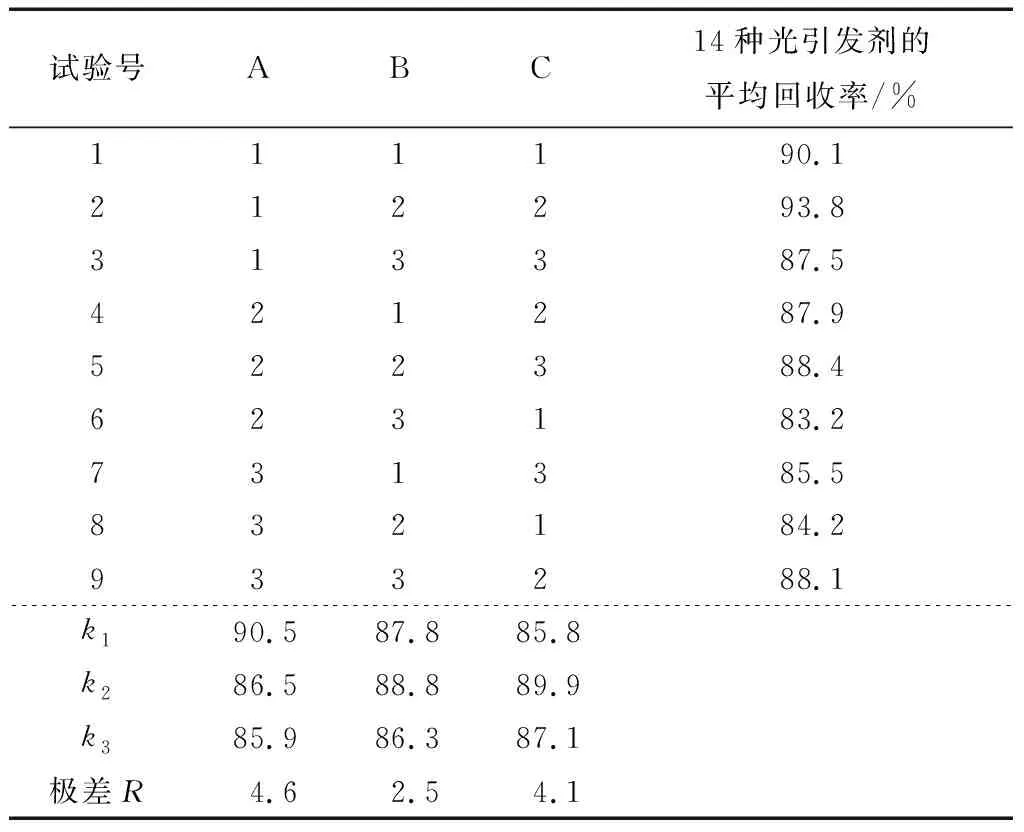
表5 正交試驗結果與極差分析Table 5 Orthogonal experiment result
2.3.2 過柱體積 由于固相萃取柱容積的局限性,需要在盡可能對樣品體積進行濃縮的前提下保證萃取效率。因此,試驗對由丙酮—正己烷提取后的上清液在進行HLB過柱凈化時的溶液體積也進行了考察。分別選用15,30,50 mL待凈化溶液過柱,考察不同體積對待測化合物回收率的影響。當過柱體積為30 mL時,所有的待測物回收率處于3種水平中的最高值。當溶液為50 mL時,所有的待測物回收率均有所下降。而當待凈化溶液為15 mL時,米氏酮和四甲基米氏酮的回收率相比30 mL時的下降最為明顯,達到了30%以上。
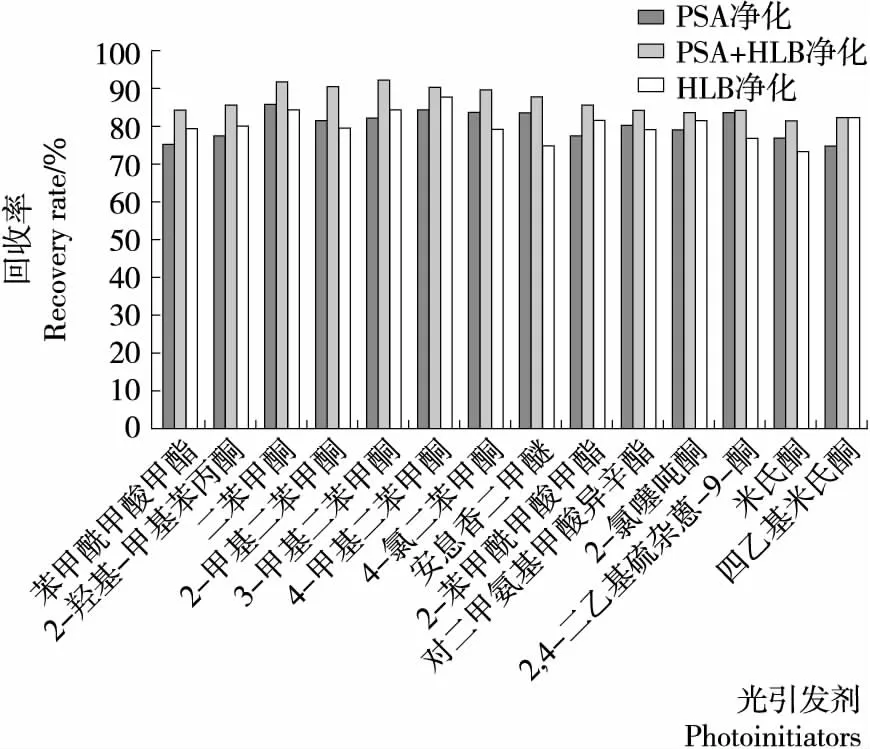
圖5 3種凈化方法對樣品中14種光引發劑加標回收率的影響
圖5 Effects of three purification methods on the recoveries of 14 photoinitiators in the spiked samples
2.4 回收率和精密度
為了驗證本方法的準確度和精密度,選取了3個有代表性的濃度做加標試驗,進行6次平行樣測試,方法的回收率和精密度見表6。由表6可知,RSD均<10%,在3個濃度下的加標回收率均滿足GB/T 27404—2008 對方法確認技術參數的要求。本試驗中14種光引發劑的回收率為73.0%~92.4%,回收率的相對標準偏差為4.1%~8.9%。
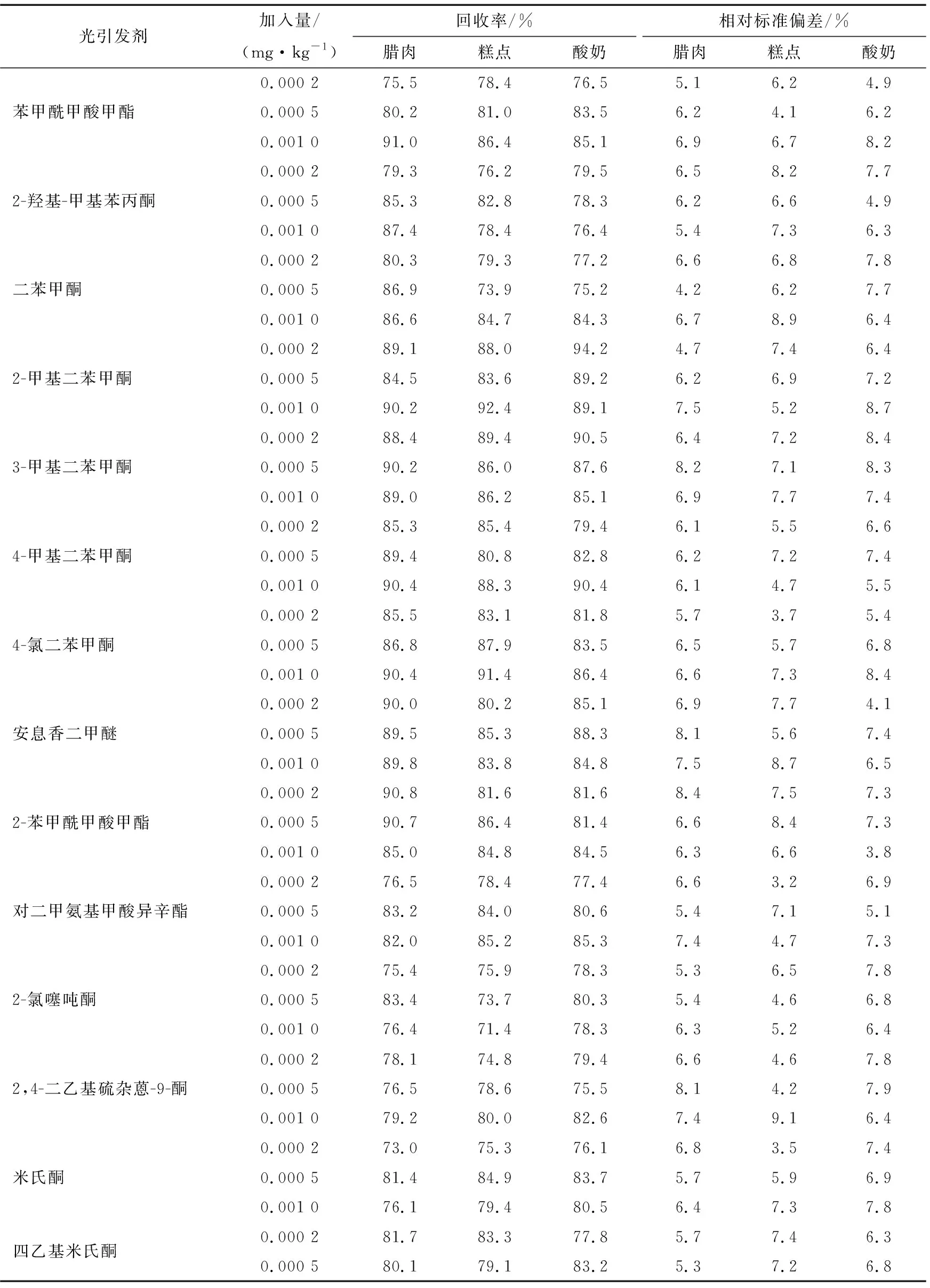
表6 14種光引發劑的加標回收率及精密度Table 6 The recoveries and RSD of 14 photoinitators
2.5 實際樣品檢測
采用本方法對40個樣品(代表基質為酸奶、牛奶、臘肉和面包)進行了檢測,共有2個牛奶樣品分別檢測出了二苯甲酮和2-氯噻噸酮,其他樣品中均未檢出光引發劑。
3 結論
本試驗建立了氣相色譜—質譜聯用法同時測定高脂高蛋白食品中14種光引發劑的方法。方法采用HLB固相萃取小柱和PSA凈化管進行聯合凈化。所得的方法檢出限為0.02~0.20 μg/kg,14種光引發劑的回收率為73.0%~92.4%,回收率的相對標準偏差均小于10%,方法的檢出限低,重現性好,準確度較高。從方法前處理到檢測結果的確認少于3 h,實現了14種光引發劑在此類食品中的快速高效檢測。下一步將圍繞食品類別與光引發劑檢出及含量關系進行研究,并擴大食品類別及光引發劑的種類對方法的適用性進行進一步評估。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
