手性2,2'-聯吡啶衍生物的不對稱合成
2018-08-28 11:20:12王廣慧喬秀麗遲彩霞董立強
綏化學院學報 2018年9期
蘇 適 王廣慧 喬秀麗 遲彩霞 王 斌 董立強
(綏化學院食品與制藥工程學院 黑龍江省綏化 152061)
腫瘤血管是腫瘤生長和轉移的病理基礎,血管內皮生長因子是腫瘤血管生成的重要調節因子,它與腫瘤血管的生成有非常密切聯系。缺少新生血管的人體腫瘤會一般被限制在很小的范圍內無法生長擴散,因此,抑制和阻斷血管內皮生長因子在腫瘤組織中的表達可達到抑制腫瘤細胞的生長和轉移的目的[1,2]。研究結果顯示,分子中含有氮芳雜環、酰胺結構以及亞胺結構的血管內皮生長因子小分子抑制劑,在體外細胞和動物的實驗中對腫瘤細胞均表現出良好的抑制作用。
本文對此類化合物構效關系分析發現,分子中的芳環或取代芳環可作為疏水性基團,分子中氮芳雜環中的氮原子、酰胺鍵可與血管內皮生長因子產生氫鍵作用[3]。因此,設計合成具有多個結合位點,生物活性高的血管內皮生長因子小分子抑制劑已經成為抗腫瘤藥物研究的熱點。基于對血管內皮生長因子抑制劑構效關系分析,設計了以吡啶衍生物為起始原料,通過縮合反應引入不同保護基和不同構型的光學純的α-氨基酸作為手性源來調控分子的立體化學特性,對手性聯吡啶衍生物合成路線進行設計如Scheme1,對所合成的化合物進行抗腫瘤活性進行檢測。
一、實驗部分
(一)儀器與試劑。核磁共振儀(BRUKE-400);旋光儀(PerkinElmerModel341LC);高分辨質譜儀(JMS-800D);無水四氫呋喃、乙醚使用鈉絲除水后重蒸,無水苯、甲苯、二氯甲烷使用鈉絲除水,丙酮采用分子篩除水。
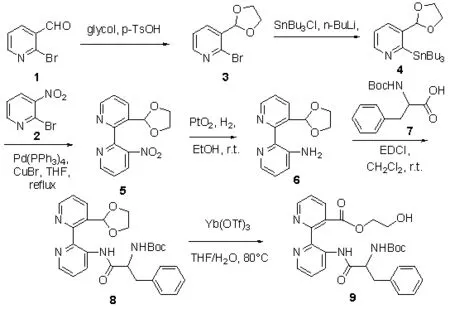
Scheme 1
化合物(3):2-溴-3-(1,3-乙二醇縮醛-2-烷基)吡啶按文獻方法合成[4],其余試劑均為分析純,其中無水四氫呋喃、乙醚使用鈉絲除水后重蒸,無水苯、甲苯使用鈉絲除水。
(二)化合物的合成。化合物(4):(三丁基錫)-3-(1,3-乙二醇縮醛-2-烷基)吡啶的合成。氮氣保護下,將(3)(1.56g,0.68mmol,1.0equiv)溶于 3.0mL無水乙醚,在 -70℃下逐滴加入正丁基鋰(0.96mL,1.6M),在-70℃下攪拌30min。用雙針頭管將溶于1mL無水乙醚的正三丁基氯化錫0.66mg(2.04mmol)轉移至反應液中,在-70℃下攪拌0.5h。TLC檢測反應結束后,加入水淬滅反應,用二氯甲烷萃取,飽和碳酸氫鈉水洗,無水硫酸鈉干燥有機相,減壓蒸除溶劑,柱層析分離純化(V(乙酸乙酯):V(石油醚)=1:40)蒸發溶劑得(4),無色油狀物,收率 90;1HNMRδ:(dd,J=4.8Hz,2.0Hz,1H),7.73(dd,J=8.0Hz,1.4Hz,1H),7.10-7.20 (m,1H),5.7(s,1H),3.90-4.20(m,4H),1.50-1.70(m,6H),1.25-1.4(m,6H),1.15-1.25(m,6H),0.80-0.95(m,9H)
化合物(5):2-[3-(1,3-乙二醇縮醛-2-烷基)吡啶]-3-硝基吡啶的合成。在氮氣的保護下,將(2)105mg(0.238 mmol)和(4)72.1mg(0.357mmol),溶于 7mL無水四氫呋喃中,加入 27.5mg(0.024mmol)三苯基磷鈀,2.7mg(0.019 mmol)溴化亞銅,加熱回流4.5h。TLC檢測反應結束后,冷卻反應液,減壓蒸除去溶劑,柱層析分離純化((乙酸乙酯):V(石油醚)=1:40)蒸發溶劑得(5),黃色油狀物,收率85%;1H NMRδ:dd,J=4.6Hz,1.4Hz,1H),dd,J=4.8Hz,1.6Hz,1H)dd,J=8.2Hz,1.4Hz,1H),dd,J=7.8Hz,1.4Hz,1H),7.55 (dd,J=8.4 Hz,4.8Hz,1H),7.42(dd,J=8.0Hz,4.8Hz,1H),6.10(s,1H),3.80-3.95(m,4H);13CNMRδ:(154.0,153.1,152.0,149.4,145.8,135.6,132.6,132.4,132.1,132.0,131.9,128.6,128.5,123.6,123.5,100.5,65.1(2C);HRMS(ESI)calculated forC13H12N3O4[M+H]+:274.0822,found274.0828(-1.9ppm,-0.6mmu)
化合物(6):2-[3-(1,3-乙二醇縮醛 -2-烷基)吡啶]吡啶-3- 胺的合成。將(5)52.9mg(0.194mmol)溶于 8mL乙醇,加入 PtO2·3H2O5.45mg(0.019mmol),1atm 的氫氣壓力下催化氫化,反應進行4.0h,TLC檢測反應結束。經過硅藻土過濾懸浮物,減壓蒸除溶劑,柱層析分離純化(V(乙酸乙酯):V(石油醚)=1:40)蒸發溶劑得(6),黃色半固體,收率 94%;1HNMRδ:(dd,J=4.8Hz,2.0Hz,1H),8.13(dd,J=8.0Hz,2.0Hz,1H),8.07-8.10(m,1H),7.60-7.70(m,1H),7.40-7.5(m,1H),7.34(dd,J=8.0Hz,4.8Hz,1H),4.60-4.8 (br,NH2),4.05-4.(m,2H),3.90-4.00 (m,2H);13C NMRδ:(156.5,148.5,142.4,141.2,138.6,133.6,132.1,132.0,131.9,128.6,128.5,124.0,123.8,122.9,100.3,65.5;HRMS(ESI)calcdlated forC13H14N3O2[M+H]+:244.1081,found 244.1089(-3.7ppm,-0.8mmu)
化合物 8(S)和 8(R)的合成通法。將(6)97.6mg(0.4 mmol)和(7)(60mmol)溶于 10mL 二氯甲烷中,加入 EDCI 191.7mg(0.6mmol),室溫下攪拌,TLC 檢測反應結束后,水洗(3×10mL),無水硫酸鈉干燥有機相,過濾,減壓蒸除溶劑,柱層析法分離純化,得純品。
8(S):無色蠟狀固體,產率 80%,aD23=-24.4(c=2.5,CHCl3);1HNM(CDCl3):(br,1H),8.84(s,1H),8.68(s,1H),8.44(s,1H),8.3(d,J=2.8Hz,1H),7.40-7.50(m,1H),7.30-7.39(m,1H),6.4(s,1H),4.95(s,1H),4.30(s,1H),3.90-4.20(m,4H),1.60-1.9(m,2H),1.20-1.6(m,10H),0.93(d,J=6.4Hz,6H);13CNMR(CDCl3):(171.7,155.5,155.3,147.7,144.2,143.5,137.4,134.7,134.1,129.2,123.7,123.3,100.2,80.1,65.4,54.2,42.0,29.7,28.3,24.9,23.0,21.8;HRMS(ESI) calculated forC24H33N4O5[M+H]+:457.2445,found 457.2452(-1.4ppm,-0.7mmu)
8(R):白色固體,產率 47%,aD23=-40.8(c=4.0,CHCl3);1HNMR (CDCl3):(br,1H),8.83 (d,J=7.6Hz,1H),8.68(s,1H),8.40-8.48(m,1H),8.21(d,J=7.6Hz,1H),7.42(dd,J=7.6Hz,4.8Hz,1H),7.32(dd,J=8.6Hz,4.6Hz,1H),6.42(s,1H),4.95 (d,J=7.2Hz,1H),4.27 (d,J=2.8Hz,1H),3.90-4.15(m,4H),1.60-1.80 (m,2H),1.20-1.50 (m,10H),0.95(d,J=6.4 Hz,6H);13CNMR(CDCl3):(171.6,155.5,147.7,144.2,143.5,137.4,134.7,134.2,129.2,123.7,123.3,100.2,80.1,65.4,54.2,42.1,29.7,29.6,28.3,24.9,23.0,21.8;HRMS(ESI)calculated for C24H33N4O5[M+H]+:457.2445,found457.2454(-1.9ppm,-0.9mmu)
化合物 9(S)和 9(R)的合成通法。將(8)(50.0mg,0.1 mmol,1.0equiv)溶 于 THF-H2O(10mL,4:1v/v)Yb(OTf)3(76.15mg,0.12mol,1.1equiv) 室溫下攪拌,TLC 檢測反應結束后,二氯甲烷萃取反應液(3×10ml),無水硫酸鈉干燥有機相,過濾,減壓蒸除溶劑,快速柱層析法分離純化,得純品。
9(S):無色蠟狀固體,產率 70%,aD23=-40.3(c=3.0,CHCl3);1HNMR(CDCl3):((br,1H),8.98(d,J=7.6Hz,1H),8.77 (s,1H),8.34 (dd,J=4.8Hz,1.2Hz,1H),8.02(d,J=7.2Hz,1H),7.30-7.60 (m,2H),4.60-5.10 (m,1H),4.10-4.60(m,3H),2.78 (d,J=13.6Hz,2H),2.20 (s,OH),0.90-1.99(m,18H);13CNM(CDCl3):(172.3,168.9,155.5,148.2,142.9,140.8,137.7,135.1,132.1,132.0,130.3,129.2,128.6,128.4,124.7,122.5,80.2,69.8,67.2,64.3,63.7,60.6,55.0,54.5,42.0,29.6,29.4,28.3,25.3,24.9,23.0,21.8;HRMS(ESI)calculated for C24H33N4O6[M+H]+:473.2395,found 473.2407(-2.7ppm,-1.2 mmu)
9(R):無色蠟狀固體,產率 55%,aD23=+41.1(c=2.6,CHCl3);1HNMR(CDCl3):(br,1H),8.99(d,J=6.4Hz,1H),8.77(s,1H),8.35(dd,J=4.4Hz,1.6Hz,1H),8.02(d,J=6.8Hz,1H),7.30-7.50(m,2H),4.99(d,J=7.2Hz,1H),4.10-4.60(m,3H),3.80 (s,2H),2.22 (br,OH),0.90-1.95(m,18H);13CNMR(CDCl3):(172.3,169.0,155.5,155.3,148.1,142.9,140.8,137.6,135.1,130.4,129.2,124.7,122.5,80.2,67.3,60.7,60.4,54.5,42.1,29.7,28.3,24.9,23.1,22.6,21.8;HRM (ESI)calculatedforC24H33N4O6[M+H]+:473.2395,found473.2404(-1.9ppm,-0.9mmu)
(二)體外抗腫瘤活性測試。活性測試方法:將細胞接種于細胞培養板上,根據靶細胞不同選擇每孔104~106個不同的細胞數。加入待測的化學藥物,同時設相應的對照,置于37℃,5%CO2環境中培養,培養時間隨測定內容及靶細胞不同而異。培養終止前4h加入5mg/mL的MTT溶液10~25ul/孔。終止培養,將所形成的結晶物用有機溶劑完全溶解后,在分光光度計上測定其再波長490nm或570nm處得A值。統計獲得的實驗數據,通過IC50計算軟件,自動擬合生成曲線圖活性因子的單位、效應細胞殺傷率等。
二、結果與討論
(一)化合物的合成。化合物(8)的合成過程中,在還原產物(6)中引入不同光學純的α-氨基酸作為手性源,反應中偶聯劑DCC難以完全除盡,在1H NMR譜的高場有雜質峰存在,影響化合物結構鑒定。選用易于水洗除去的EDCI作為偶聯劑,能夠克服上述缺點。
(二)體外抗腫瘤活性。選擇五種不同的腫瘤細胞株(HCT-8:人結腸癌細胞;Bel7402:人肝癌細胞;BGC-823:人胃癌細胞;A549:人肺腺癌細胞;A2780:人卵巢癌細胞)對目標化合物8和9的抗腫瘤活性進行了生物評價,見表1。

表1 目標化合物抗腫瘤活性篩選
評價數據結果表明,目標分子對所選用的五種腫瘤細胞株的增殖沒有明顯的抑制活性。隨后,為了改變化合物的酯水分配系數,提高其跨膜能力,增強化合物與血管內皮生長因子的作用能力,對化合物8和9進行了化學修飾和結構改造。期待通過對化合物8和9結構的改造來調整化合物的酯水分配系數,提高分子的跨膜能力,有利于分子進入腫瘤細胞內部與血管內皮生長因子發生相互作用,提高化合物的抗腫瘤增殖的能力,期望發現與血管內皮生長因子具有高親和力的新型先導化合物。
三、結論
本文設計并合成了一系列新型的含有手性中心的聯吡啶類化合物,其結構經1H NMR,13C NMR和HRMS譜圖確證,實驗結果該方法對合成具有潛在生物活性的手性聯吡啶化合物的有一定借鑒價值。MTT實驗結果顯示,目標化合物對 HCT-8、Bel7402、BGC-823、A549、A2780 細胞無顯著抑制活性。
