皮下脂膜炎樣T細胞淋巴瘤的臨床分析
2018-08-24 03:49:54劉玉峰徐學聚
實用癌癥雜志 2018年8期
張 園 劉玉峰 徐學聚
皮下脂膜炎樣T細胞淋巴瘤(SPTCL)是1種原發(fā)于皮下脂肪組織的外周 T 細胞淋巴瘤,起源于分化階段不明的細胞毒T細胞。于1991年Gonzalez首次臨床報道,臨床上較為罕見,占所有非霍奇金淋巴瘤的l%以下[1]。生長方式類似脂膜炎,獨特的臨床表現(xiàn)與其它類型的外周T細胞淋巴瘤相區(qū)別。本病好發(fā)于中青年,兒童罕見。臨床上主要表現(xiàn)為多發(fā)對稱、紅或褐色質硬的皮下結節(jié),好發(fā)于四肢和軀干,少見于面部。60%的病例伴有體重減輕、發(fā)熱等全身癥狀,可伴有肝脾及淋巴結腫大。臨床病程常反復遷延,早期治療預后較好,病程發(fā)展速度較快,5年生存率可高達80%以上[2]。由于分類的縮改變,區(qū)別于2008年以前報道的SPTCL預后存在很大差異。為探討更有效地SPTCL的治療手段,筆者選擇近8年內6例本院病理及免疫學診斷為SPTCL的臨床病例進行分析,結果分析如下。
1 資料與方法
1.1 資料
收集鄭州大學第一附屬醫(yī)院病理科2009年01月至2016年10月診斷并經(jīng)2名病理醫(yī)師復檢,且有完整隨訪資料的SPTCL病例6例。活檢組織標本由北京市淋巴腫瘤會診中心依據(jù)2008年世界衛(wèi)生組織(WHO)出版的"淋巴造血組織腫瘤分類"進行診斷[3]。對其臨床表現(xiàn)、組織病理形態(tài)、免疫表型、組織起源及治療后隨訪進行分析總結。應用病理免疫表型檢測T淋巴細胞標志CD3陽性,B淋巴細胞標志CD20陰性。免疫組化進一步支持腫瘤細胞為T淋巴細胞來源。分期根據(jù)Ann Arbor標準分期。年齡11~50歲,平均年齡28.6歲;男性5例,女性1例。
1.2 臨床表現(xiàn)
皮膚病變部位包括四肢、面部、頭皮、軀干、眼瞼、臀部。6例以全身多發(fā)紅斑、皮下結節(jié)為首發(fā)癥狀,病變處皮溫增高,多有腫脹、疼痛,也可無自覺癥狀,無特異性形狀,單發(fā)或多發(fā),大小不一,部分表面出現(xiàn)潰瘍。5例伴淋巴結腫大,5例伴隨發(fā)熱,1例伴有脫發(fā),2例骨髓侵犯,1例出現(xiàn)肝功能異常。2例患者為Ⅲ/Ⅳ期,4例患者為Ⅳ/Ⅳ期。2例治療前乳酸脫氫酶(LDH)均高于正常。均未合并噬血細胞綜合癥(HPS)癥狀,1例兒童初診時骨髓可見吞噬現(xiàn)象,臨床達不到噬血細胞綜合癥臨床標準。多數(shù)病例伴有全身癥狀:發(fā)熱、乏力、肌痛、關節(jié)痛,有不同程度血常規(guī)異常。6例病例臨床特征見表1。
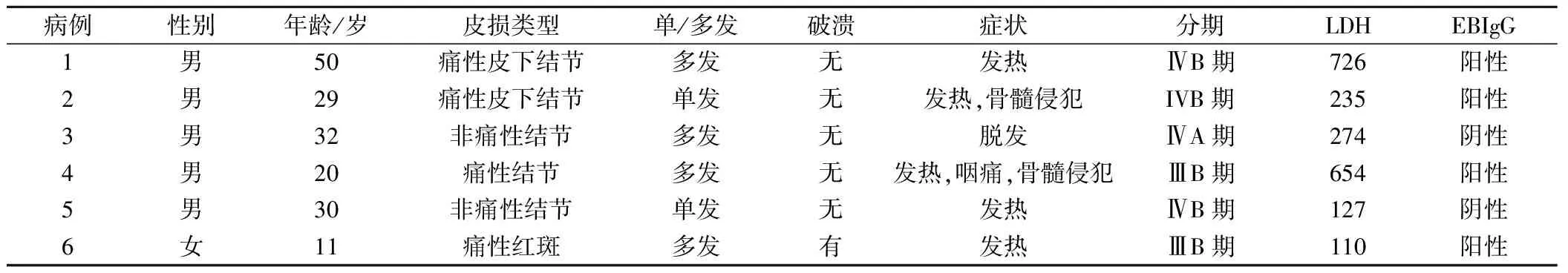
表1 6例SPTCL患者臨床表現(xiàn)
1.3 病理學及免疫學特點
病理學特征:皮下脂肪組織中大小不一的非典型性淋巴細胞浸潤,鏡下淋巴瘤細胞花環(huán)狀圍繞單個脂肪細胞排列,中等或大細胞,染色質濃染,常見核分裂。病理組織免疫學特點:選擇抗體CD3、CD4、CD8、CD56、TIA-1、GranzymeB、CD45、CD68、CD20、TIA-1、βFl、CD79a,采用EnVision二步法免疫組織化學染色標記,以已知陽性切片作為陽性對照。PBS緩沖液代替一抗作空白對照。病理組織標本應用EB病毒編碼的小分子寡核苷酸(EBER)原位雜交方法。選用異硫氰酸熒光素(FITC)標記的針對EBER探針EBERl/2在石蠟切片上做原位雜交,進行顯色分析,光鏡觀察,以EBERl/2陽性鼻咽癌的石蠟切片作為陽性對照。DEPC水代替含探針的雜交液為空白對照。6例基因檢測均可見TCR基因重排。6例免疫學特征見表2。
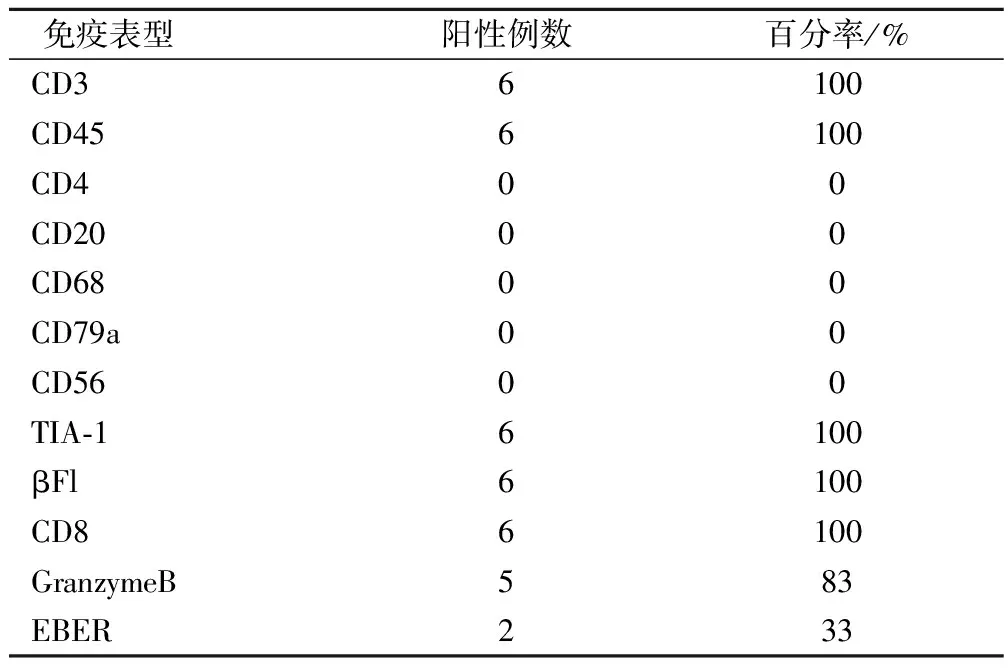
表2 相關抗原陽性率
1.4 療效判斷標準
按WHO標準分為完全緩解(CR),部分緩解(PR),穩(wěn)定(SD),進展(PD)。隨訪時間自治療開始到死亡或末次隨訪時間。
2 結果
6例患者治療方案主要為化療,其中包含ECHOP、CHOP、MINE、GMED、DDGP、GDPT、ESHAP。1例患者聯(lián)合放療,1例化療復發(fā)后選擇自體干細胞移植。治療過程及治療效果,見表3。

表3 6例SPTCL患者治療過程及預后
注:DDGP:吉西他濱,順鉑,培門冬酶,強的松,CHOP:多柔比星,潑尼松,長春新堿,環(huán)磷酰胺,ECHOP:依托泊苷,多柔比星,潑尼松,長春新堿,環(huán)磷酰胺,MINE:米托蒽醌,異環(huán)磷酰胺,依托泊苷,GMED:異環(huán)磷酰胺,依托泊苷,甲氨蝶呤,長春新堿+地塞米松,ESHAP:依托泊苷,順鉑,阿糖胞苷,強的松,GDPT:順鉑,吉西他濱,沙利度胺,地塞米松。
3 討論
SPTCL屬于外周T細胞淋巴瘤,男性發(fā)病高于女性[4],兒童發(fā)病少見。本院6例患者,平均年齡28.6歲,男性多見(5例/6例),和報道相符,兒童病例僅1例。具體發(fā)病原因目前尚不清楚,以往報道與EB病毒的感染相關,而且 EB 病毒相關的 SPTCL 發(fā)展更為迅速,可導致患者的早期死亡。近期外文文獻報道與EB病毒相關性不大,也有文獻報道對不同地域人種,EB病毒的致癌機制不盡相同,亞裔人種的相關性高于歐洲人種[5]。本臨床分析通過測定EBER(33%病例陽性)及目前EBIgG(66%病例陽性),不足以說明EB病毒的感染與SPTCL相關。
SPTCL的臨床表現(xiàn)類型多樣[6-7],常表現(xiàn)為紅色至褐色皮下結節(jié)、紅斑、潰瘍,單發(fā)或多發(fā),可伴腫脹、疼痛,皮溫升高,也可無自覺癥狀。以四肢及軀干部多見,少見于面部、頸部,結節(jié)可發(fā)展為潰瘍。皮下結節(jié)經(jīng)常因特征多樣誤診為風濕類疾病或良性脂膜炎[8]。本院收集病例1例發(fā)病面積波及面部,其余5例均為四肢或軀干。多見伴發(fā)癥狀為發(fā)熱、消瘦、乏力、肝脾淋巴結腫大、關節(jié)痛、肌痛、消瘦。在2005年新版的皮膚淋巴瘤世界衛(wèi)生組織-歐洲癌癥治療研究組織(WHO-EORTC)分類中已明確限定SPTCL為 TCRа/β +型[9]。隨著診斷界限的改變,合并噬血細胞綜合癥(HPS)較以往文獻報道存在明顯減少[10]。6例病例有不同程度血常規(guī)異常,均未合并噬血細胞綜合癥癥狀,1例兒童初診時骨髓可見吞噬現(xiàn)象,臨床達不到噬血細胞綜合癥臨床標準。
皮膚活檢組織的特征性病理及免疫組化是診斷SPTCL的主要方法,聯(lián)合分子生物學的支持對該病進行診斷。皮膚病變大多局限于皮下組織,為小葉內脂膜炎樣浸潤,幾乎無真皮及表皮損害。鏡下淋巴瘤細胞花環(huán)狀圍繞單個脂肪細胞周圍,并浸潤一些小靜脈,但無血管破壞。可見組織細胞吞噬紅細胞和壞死碎屑形成特征性豆袋細胞,可見散在的核碎片及脂肪壞死[11]。特征性免疫表型為CD3+、CD8+、CD4-、CD56-、βFl+、部分表達細胞毒性顆粒蛋白TIA-1。其中βFl是1種與T細胞表面受體,β鏈非多肽框架決定簇結合的單克隆抗體,該抗體陽性可證實為TCRа/β+型淋巴瘤,區(qū)分于以往的SPTCL分類。在2008年的WHO惡性淋巴瘤分類中被確定為1種獨立的類型。將SPTCI僅限定于TCRа/β+型,而將TCRγ/σ+型重新命名為原發(fā)皮膚γ/σ+T細胞淋巴瘤。PCR可檢測到TCR基因重排。因基因重排陽性可見于淋巴細胞增生性良性疾病,故只用于對SPTCL診斷的支持,陰性結果不可排除SPTCL。6例病例均在病理及免疫組化支持SPTCL的診斷,6例基因檢測均可見TCR基因重排。
皮下脂膜炎樣T細胞淋巴瘤治療手段包括:單藥治療、聯(lián)合化療、手術切除、生物治療、放療、造血干細胞移植。①單藥治療:包括應用糖皮質激素,免疫抑制劑,血管生成抑制劑治療。有報道早期口服激素可使進展緩慢單發(fā)皮損的患者腫瘤病變達到長期臨床緩解,殘留檢查仍可見淋巴瘤細胞[12]。仍有少量病例在早期誤診為風濕性疾病,應用激素后,病情可暫時控制,皮下結節(jié)仍反復出現(xiàn)。在治療淋巴瘤中,激素的療效在臨床上肯定,應用于在SPTCL的急性期或早期。化療療效不佳的患者,免疫抑制劑環(huán)孢素可迅速控制疾病進展,達到較好的效果。對于復發(fā)難治的SPTCL患者,或不適合行放療及移植治療的患者,可使用血管生成抑制劑沙利度胺或較新的藥物如硼替佐米[13]。沙利度胺可以活化T細胞及NK細胞并增加NK 細胞的數(shù)量,抑制血管內皮生長因子(VEGF)來抑制血管新生等作用機制來改變腫瘤所處的微環(huán)境,從而起到抗腫瘤作用,近年來也開始被用于SPTCL的治療[14]。在本院6例病例中,主導方案早期均聯(lián)合應用糖皮質激素,3例病例晚期均應用沙利度胺,最終4例在隨訪時間內達到緩解,1例死亡1例復發(fā)。②聯(lián)合化療:在臨床的應用最為廣泛,SPCTL在臨床上病例數(shù)目較少,治療效果在既往未界定于TCRа/β+型時預后較差,而且很多方案均具有局限性,或存在較大毒副作用,或存在年齡依賴等因素,無明確療效的固定化療方案。一線治療方案常應用淋巴瘤CHOP或其他蒽環(huán)類藥物為基礎的方案,Ronald等回顧性分析的156 例SPTCL 患者中有38 例患者最初治療選擇CHOP方案,總有效率(OR)為53%[15]。二線化療方案包括持續(xù)EPOCH、CHOP、MINE、GMED、DDGP、GDPT、ESHAP。吉西他濱為主的化療方案對T細胞淋巴瘤效果確切,其促進腫瘤細胞凋亡的機制包括競爭性抑制合成DNA的脫氧胞苷酸、核糖核酸還原酶,從而抑制DNA合成。在復發(fā)難治的外周T細胞淋巴瘤中較為常用。一項新的長期隨訪臨床研究報道認為,吉兩他濱應被作為首選治療藥物[16]。文獻報道CMED方案在遠期生存率方面遠優(yōu)于CHOP,且毒性反應輕微,患者多數(shù)可以耐受[17]。目前報道的聯(lián)合化療方案,鮮有多中心大樣本臨床試驗,且目前尚未得到廣泛的學者公認,需要進一步的臨床試驗來確認療效。本院6例患者治療均以聯(lián)合化療為主導,1例兒童患兒治療早期應用基礎的CHOP化療方案,最后復發(fā)。5例成人患者運用聯(lián)合化療均選用以吉西他濱為主的化療方案,3例完全緩解(CR),1例復發(fā)后應用BEAM預處理方案后行自體干細胞移植(autoHSCT)最終緩解,1例進展(PD)后應用(博來霉素+長春新堿)后骨髓抑制合并感染后死亡。③手術切除:切除病變部位也是姑息治療方案中1種,用于不能耐受化療方案且病變位置局限的少數(shù)病例,國外文獻報道療效較差,復發(fā)率較高[18]。本院6例病例未應用手術切除,無法證明手術切除對SPTCL的療效。④生物治療:除外傳統(tǒng)治療方法,生物治療也被越來越多的使用,如貝沙羅汀,1種口服維甲酸類似物。在外文文獻中極少數(shù)病例應用,療效有待評估。⑤放療:患者就診時多處于晚期(Ⅲ或者Ⅳ期),放射治療效果欠佳,副作用較大,故臨床上很少對SPTCL患者進行放射治療。Leiteh等的研究表明,放射治療能明顯影響早期SPTCL患者的無進展生存率(PFS)。但是是否能夠改善患者的生存時間還有待更多試驗進一步證實。據(jù)Go統(tǒng)計,單獨應用放療,完全緩解率為36%(4/11),部分緩解為45%(5/11),總緩解率為81%(9/11),緩解期從12月到23個月不等。放療同時應用系統(tǒng)治療,包括聯(lián)合化療及潑尼松,干擾素治療,幾乎所有的皮損,均可局限。放射免疫治療(RIT)是近幾年1種新興的治療SPTCL的方法,是將放射性核素與CD20單克隆抗體(如托莫西單抗)進行藕連,將放射性核素導向腫瘤部位,而周圍正常組織的照射量很少,從而起到局部治療作用。本院僅1例成人患者聯(lián)合放療,最終死亡。⑥造血干細胞移植:大劑量化療和造血干細胞移植在復發(fā)或難治性SPTCL病例中仍可獲得較高CR率,機制尚不明確。也有學者報道用異體外周血干細胞移植(allogeneic peripheralblood stem cell transplantation,allo-PBSCT)或自體干細胞移植治療難治性SPTCL,可完全緩解[19]。外文文獻中單中心小樣本回顧性分析,13例接受干細胞移植,其中12例為自體干細胞移植,1例為異體干細胞移植,均獲得完全緩解,其中進行異體干細胞移植的病例完全緩解期最長,達70個月,而其他自體干細胞移植病例完全緩解持續(xù)時間中位數(shù)為14個月,且有1例接受干細胞移植3個月后復發(fā)[20]。本院僅1例復發(fā)后應用BEAM預處理方案后行自體干細胞移植(autoHSCT)最終緩解,療效明確,可應用于難治性或復發(fā)的少數(shù)病例。
由于SPTCL在WHO-EORTC分類中被歸類為惰性或低度惡性皮膚T細胞淋巴瘤,其病程可遷延反復長達多年,甚至數(shù)十年,預后良好。其5年生存率可達80%以上。患者若伴發(fā)噬血細胞綜合征,則在短期內死亡。SPTCL最主要的死亡原因主要為:骨髓抑制合并感染性休克、肺部感染、腦出血、彌漫性血管內凝血和休克。本院6例病例其中4例疾病緩解,1例兒童病例,應用3個療程的ECHOP,出現(xiàn)發(fā)熱,再次皮下結節(jié)出現(xiàn),應用GMED部分緩解。3例初始治療應用CHOP或ECHOP均效果差未達到持續(xù)緩解,應用含吉西他濱方案治療初治和復發(fā)患者5例,3例完全緩解(CR),1例復發(fā)后應用BEAM預處理方案后行自體干細胞移植(autoHSCT)最終緩解,1例進展(PD)后應用(博來霉素+長春新堿)后骨髓抑制合并感染后最終全身多臟器功能衰竭死亡。Gllamini等報道了1個多中心的 385例SPCTL的回顧性臨床研究,提出了預后的危險因素包括年齡>60歲,血清乳酸脫氫酶(LDH)增高,有骨髓侵犯,IPI>2分[21]。6例患者復發(fā)及死亡2例,均有骨髓侵犯,且血清LDH增高。6例IPI均≤2分,年齡均≤60歲,基于病例數(shù)目較少,不能完全支持以上觀點。國內報道,初次化療是否能達到CR、臨床分期、B癥狀相關亦是預后因素,但是尚未在臨床推廣應用。依靠6例病變過程及轉歸,我們得出:一旦確診SPTCL,應立即給予患者早期給予高強度、大劑量化療,以控制病情,晚期維持給予單藥化療,以減少復發(fā)。
