非貴金屬單原子催化劑的研究進展
2018-08-10 09:47:44吳耿煌榮峻峰達志堅
石油學報(石油加工) 2018年4期
關鍵詞:催化劑
吳耿煌, 黃 凰, 榮峻峰, 達志堅
(1.中國石化 石油化工科學研究院, 北京 100083; 2.國民核生化災害防護國家重點實驗室, 北京 102205)
催化劑作為催化技術的核心,能夠有效地調控化學反應的路徑,在更為溫和的條件下,高選擇性地獲得目標產品[1]。工業催化中催化劑與反應物通常處于不同的相態,因此催化過程主要涉及多相催化。其中,負載型金屬催化劑由于具有較高的催化活性與選擇性,被廣泛應用于重要的化工產品的生產過程。對于負載型金屬催化劑,金屬顆粒的尺寸是影響其催化性能最重要的因素之一[2,3]。表面原子與總原子數之比隨著粒徑的變小而急劇增大,產生了更多的配位不飽和金屬中心。此外,當顆粒尺寸下降到一定值時,費米能級附近的電子能級由準連續能級變為分離能級,產生量子尺寸效應;同時粒徑的減小還增強顆粒與載體之間的相互作用,這都直接影響到催化劑的性能[4-6]。直觀上看,為了最大限度地利用金屬材料,將金屬以單原子的形式分散在載體表面并且作為活性位點,是多相催化所能達到的極限[7-9]。
2011年,張濤院士團隊利用氧化鐵與鉑原子之間的強相互作用,成功制備出首例具有高活性和高穩定性的單原子鉑催化劑Pt1/FeOx[10],引發了國際上對單原子催化劑的研究熱潮。單原子催化劑是指催化劑中活性組分完全以孤立的單個原子的形式存在,并通過與載體作用或與第二種金屬形成合金得以穩定。相比于納米/亞納米催化劑,單原子催化劑具有諸多優勢:(1)最大限度地提高金屬原子利用率;(2)活性位點的組成和結構單一,可避免因活性組分組成和結構不均勻導致的副反應;(3)兼具均相催化劑均勻單一的活性中心和多相催化劑結構穩定易分離的特點,因此單原子催化劑有望成為連接均相催化與非均相催化的橋梁[11,12]。繼Pt1/FeOx的成功制備之后,高性能的Ir/FeOx[13],Au/CeO2[14]和Pd/ZnO[15]等貴金屬單原子催化劑的研究也取得了突破。然而考慮到貴金屬的稀缺性,發展可能替代貴金屬催化劑的高性能非貴金屬單原子催化劑顯然具有更大的價值空間。在自然界,固氮酶中的Mo,血紅素中的Fe,葉綠素中的Mg等都可以看成是高效的非貴金屬單原子催化劑[16]。最新的研究還表明,基于Fe、Co等的非貴金屬單原子催化劑在某些催化反應中所體現的性能足以媲美甚至優于貴金屬催化劑[17,18]。鑒于非貴金屬單原子催化的研究近年來取得諸多進展及巨大的發展潛力,在本文中,筆者重點介紹了非貴金屬單原子催化劑的制備、表征方法及其在電催化、加氫及脫氫反應、液相氧化反應、光催化還原CO2等領域的應用研究進展,同時展望了非金屬單原子催化劑的發展趨勢。
1 非貴金屬單原子催化劑的制備方法
1.1 原子層沉積法
原子層沉積(ALD)也稱為原子層外延,是一種將氣相前驅體脈沖交替地通入反應腔并在基體表面發生化學吸附及化學反應,形成單原子沉積膜的方法。ALD具有沉積參數高度可控、優異的沉積均勻性和一致性等優點,被廣泛應用于納米材料的制備[19]。Li等[20]以熱穩定性良好的Zr基金屬有機框架(MOF)NU-1000為沉積載體,雙(N,N′-二-叔-丁基乙脒基)鎳為前驅體,在NU-1000的Zr6位點上均勻沉積了原子級別分散的單位點Ni催化劑。其中Ni通過O與Zr鍵合而穩定,Ni的化學態接近二價的離子態。隨后他們又以ALD法及水熱沉積法分別在 NU-1000 上沉積了單位點的Co[21]。同單位點的Ni一樣,這兩類Co基催化劑中Co的價態均為二價離子態。此外,Qiu等[22]通過化學氣相沉積(CVD),在納米多孔Ni載體表面沉積多孔石墨烯后用HCl將Ni載體移除,殘留了少量的Ni與C直接鍵合,以單原子的形式嵌入石墨烯表面。Zhang等[23]通過水熱沉積,將Co以單位點的形式穩定在MOF-525(Zr)的卟啉環空穴中,制備了單位點MOF-525-Co催化劑。總體而言,目前各類沉積法通常需預先制備特定有序結構的載體單元,合成步驟較為繁瑣且使用的有機配體價格較為昂貴,不適用于大批量制備非貴金屬單原子催化劑。
1.2 電弧放電法
電弧放電法是一種制備碳基金屬復合材料的重要方法[24]。Zhang等[25]采用改進的電弧放電法成功制備了石墨化碳層負載單原子Nb的催化劑。圖1[25]為電弧放電法制備石墨化碳層負載單原子Nb催化劑的裝置示意圖,Nb棒為陽極,C棒為陰極,CH4為氣相C源,工作電壓30 V,工作電流90 A。需要指出的是,所制備的碳基催化劑還負載了一定量的碳包覆NbC顆粒。由于電弧放電法制備的催化劑容易產生包括純碳材料、碳包覆金屬納米顆粒等在內的雜質且不易分離,因此這類方法并不適合批量制備高純度的碳載單原子催化劑。
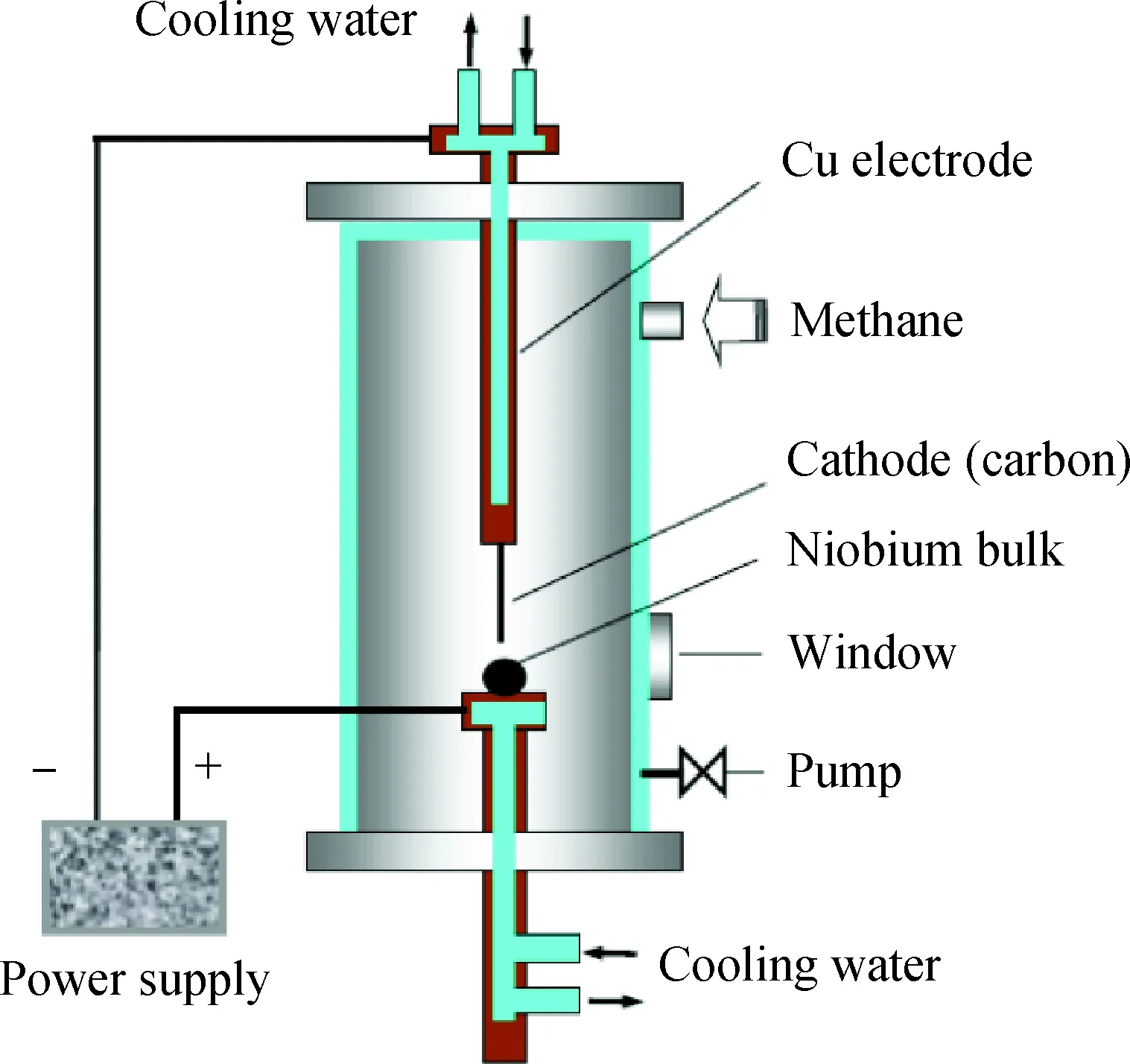
圖1 電弧放電法制備石墨化碳層負載單原子 Nb(Nb-in-C)的裝置示意圖[25]Fig.1 Schematic diagram of the synthesis equipment used to produce Nb-in-C complex[25]
1.3 高能球磨法
高能球磨法利用機械能來誘發化學反應或誘導材料組織、結構和性能的變化,以此來制備新材料[26]。Deng等[27]在Ar氣氛保護的條件下,高能球磨酞菁鐵分子與石墨烯納米片(Graphene nanosheets, GN),利用N與GN的C形成強的共價鍵,使得N作為“錨”來穩定配位不飽和的Fe中心。通過改變酞菁鐵與GN的比例,制備了Fe負載量(質量分數)分別為1.5%、2.7%以及4.0%的3種單原子催化劑。進一步利用高分辨透射電鏡(HRTEM)、高角度環形暗場掃描透射電鏡(HAADF-STEM)以及低溫掃描隧道顯微鏡等表征手段證實了Fe通過與4個N配位形成FeN4中心,并限域在石墨烯骨架中。此后他們還分別以酞菁錳、鈷、鎳、銅為金屬前驅體,成功制備了單位點的MnN4/GN、CoN4/GN、NiN4/GN、CuN4/GN,證實了以酞菁配位的非貴金屬鹽與GN為前驅體通過球磨法制備對應的N配位的非金屬單位點催化劑具有較好的普適性[28]。
1.4 液相化學還原法
液相化學還原法是指利用還原劑的還原性,在液相中將金屬陽離子還原為對應的金屬單質的方法。通過改變溶劑、還原劑、金屬前驅體、表面活性劑、載體以及反應溫度與時間,可以十分有效地調控金屬納米材料的組成、形貌與尺寸[29]。Long等[30]以TiO2納米片為載體,維生素C為還原劑,水相中還原Pd、Cu金屬鹽,將具有不同組成比例的PdxCu1合金納米顆粒負載于TiO2表面(PdxCu1-TiO2)。X射線吸收精細結構(XAFS)分析表明,當Pd/Cu摩爾比大于7時,合金中的Cu以單原子的形式與Pd成鍵,Cu之間無金屬鍵作用,并且由于Pd的保護作用,Cu以金屬態的形式穩定。盡管利用液相化學還原法通過調節金屬前驅體的比例以制備非貴金屬單原子催化劑的方法相對較為簡單,但此方法的普適性還有待進一步的驗證。
1.5 沉積-沉淀法
沉積-沉淀法是指將需負載的金屬鹽溶液與載體在攪拌條件下形成均勻的懸浮液,控制一定的溫度和pH值,使金屬沉積在載體表面,隨后進行過濾、洗滌、干燥、焙燒等處理,得到負載金屬的催化劑[31]。SiO2是最常用一類載體,其表面豐富的含O官能團可以與金屬發生鍵合,有效地穩定金屬組分[32]。Zhu等[33]采用尿素輔助的沉積-沉淀法制備了SiO2負載的單原子Cu催化劑(Cu/SiO2-UH),Cu的負載量高達15%(質量分數)。尿素在制備過程中起到2個作用,一是加熱過程中發生水解,提高溶液pH值,促進了硅醇基的去質子化;二是水解產生的NH3與Cu2+配位形成銅氨離子。銅氨離子進一步與去質子化的硅醇基發生鍵合,形成了Cu-O-Si共價鍵,實現了Cu的單原子分散。隨后用H2對催化劑進行還原處理,還原后Cu主要為Cu0以及Cu+2種價態,且HRTEM證實Cu仍為單原子分散狀態。此外將可溶性銅鹽替換成錳鹽、鈷鹽、鎳鹽和鋅鹽,也可制備出對應的SiO2負載的非貴金屬單原子催化劑。
1.6 原位活化法
原位活化法與前面所述方法的區別在于單原子催化劑是由新鮮催化劑(非單原子催化劑)在反應過程中原位活化而制得。Guo等[34]將SiO2與Fe2SiO4球磨混合后進一步高溫熔化,用HNO3瀝濾,干燥獲得了Fe?SiO2催化劑并將該催化劑應用于甲烷制乙烯、芳烴的反應中。研究發現,在1090℃的高溫下反應60 h 后,新鮮催化劑上的FeOx顆粒發生結構重構,Fe在反應過程中重新以單一位點的形式分布在SiO2基質上;XAFS進一步驗證了反應過程中Fe鑲嵌在Si 和C 之間形成了鍵合作用,保證該催化劑高溫下的穩定。此外,Fan等[35]以Ni-MOF為前驅體,經過700℃高溫碳化后,用HCl瀝濾,干燥獲得了石墨碳包覆Ni納米顆粒核殼結構催化劑并將該催化劑應用于電催化析氫(HER)反應,通過HAADF-STEM直觀地證實在電化學掃描過程中,催化劑發生原位活化,核層Ni納米顆粒逐漸溶解,以單原子的形式重新嵌入石墨化碳層中。由于原位活化法存在一定的特殊性,因此利用此方法有目的地制備非貴金屬單原子催化劑仍存在較大難度。
1.7 高溫裂解法
Co和Fe通過與碳載體上的N配位所形成的單原子分散的Co-N-C以及Fe-N-C是兩類非常重要的非貴金屬單原子催化劑[18,36]。Co(Fe)-N-C催化劑通常采用高溫裂解法制備,即將含N、C的有機配體配位的金屬前驅體與碳載體或其他模板載體在600~900℃惰性氣氛下高溫裂解碳化,是目前制備非貴金屬單原子催化劑最主要的方法。Liang等[37]以維生素B12(VB12)、聚苯胺鐵絡合物(PANI-Fe)為前驅體,SiO2納米顆粒、有序介孔的SBA-15以及層狀結構的蒙脫石3種SiO2材料為模板,經高溫裂解碳化后用HF除去模板及金屬納米顆粒,得到Co-N-C及Fe-N-C催化劑。如圖2[37]所示,通過選用不同結構的模板,可有效地調控Co(Fe)-N-C孔結構及比表面積,實現對催化劑性能的調控。Shen等[38]同樣以SBA-15為模板,浸漬FeCl3并與CCl4、乙二胺、升華S水熱混合后高溫裂解,用HF去除模板,制備了S摻雜的Fe-N-C催化劑。XAFS表明,Fe與N的平均配位數為2,主要以FeN2位點的形式負載于碳載體上。除了SiO2材料外,Mg(OH)2及MgO也可以作為一類犧牲載體,用于Co(Fe)-N-C的制備。Liu等[39]將醋酸根、菲啰啉共同配位的Co(phen)2(OAc)2負載于Mg(OH)2后,700℃高溫裂解碳化,用HNO3瀝濾MgO,干燥后獲得了質量分數高達3.6%的Co-N-C單原子催化劑。此類方法還可用于制備質量分數為1.8%的Fe-N-C單原子催化劑[40]。此外,Zhu等[41]以Te納米線為模板,制備了碳納米管(CNT)氣凝膠負載的Fe-N-C催化劑。由于Te納米線在高溫下易揮發,因此模板在高溫裂解的過程中自動移除,無需再經瀝濾、干燥等后處理步驟。除了以非碳材料作為犧牲模板外,也可直接將炭黑[42]、石墨烯[43]、CNT[44,45]以及碳鏈聚合物[46]等碳載體負載有機配體配位的金屬鹽,經高溫裂解、強酸瀝濾以制備單原子的Co(Fe)-N-C催化劑。
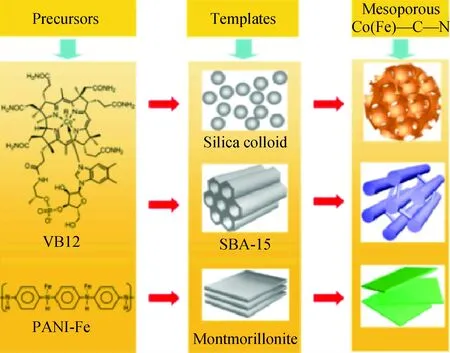
圖2 模板法制備介孔Co(Fe)-N-C催化劑的示意圖[37]Fig.2 Schematic illustration of templating synthesis of mesoporous Co(Fe)-C-N catalysts[37]
沸石咪唑酯骨架結構材料(ZIF)是一種新型的多孔晶體材料,屬于具有特定配體的MOF材料。ZIF中,有機咪唑酯通過N的配位作用,交聯連接到過渡金屬上,形成一種四面體框架[47]。由于其有序的骨架結構,既有N配位可調控的金屬中心(Fe、Co、Zn等),且配體可作為碳源,因此由ZIF制備單原子Co(Fe)-N-C催化劑具有顯著的優勢與潛力。Zitolo等[48]以負載Fe(phen)2(OAc)2的ZIF-8為前驅體,經過1050℃高溫裂解,用H2SO4瀝濾、干燥獲得了Fe質量分數為1.0%的Fe-N-C催化劑。Yin等[49]在ZIF的合成過程中同時加入Co、Zn鹽,制備了雙金屬的CoZnZIF,如圖3[49]所示,通過調控Zn與Co的比例,可有效調控ZIF中Co原子節點的距離;此外Zn在高溫裂解過程中揮發,配位的前驅體轉化為額外的N摻雜位點有利于進一步穩定Co原子,這兩種作用有效避免Co在高溫下的團聚。利用這種方法,成功制備了Co負載量>4%的單原子Co-N-C催化劑。相同的方法還可用于制備單原子Ni-N-C[50]和Fe-N-C催化劑[51]。與Co(Ni)Zn體系不同的是,FeZn體系ZIF合成的過程中Fe、Zn沒有形成雙金屬ZIF,前驅體Fe(acac)3被封裝在ZIF-8(Zn)的分子籠內。由于孔徑的限制,每個分子籠只能捕獲1個Fe(acac)3,這種特殊的結構有效避免了高溫裂解過程中Fe的團聚。此外,Zhang等[52]則合成了粒徑可控的雙金屬FeZnZIF,并經高溫裂解后,無需酸洗瀝濾,直接制備單原子Fe-N-C催化劑。
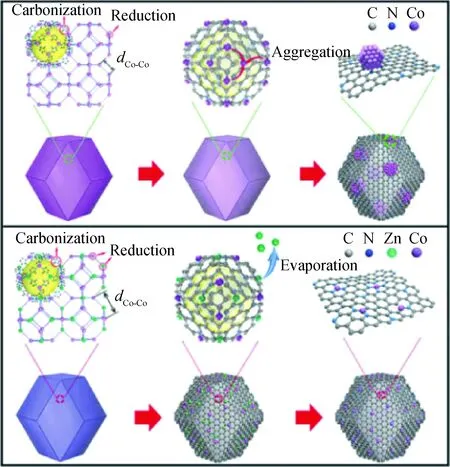
圖3 氮摻雜碳負載的Co納米顆粒(CoNPs-N/C)及 單原子Co-N-C的制備過程示意圖[51]Fig.3 Schematic illustration of the formation of Co NPs-N/C and Co-N-C[51]
最近,Zhang等[53]還提出了一種基于金屬(氫)氧化物@聚合物核殼結構制備金屬單原子催化劑的方法。如圖4所示,將FeOOH納米棒表面包覆聚多巴胺(PDA)后,經高溫碳化,HCl瀝濾,干燥后即得到單原子Fe-N-C催化劑。通過改變金屬前驅體,這種方法還可用于制備單原子的Co-N-C、Ni-N-C、Mn-N-C、FeCo-N-C、FeNi-N-C等催化劑。此外,Ohn等[54]將NiCl2與三聚氰胺的混合物高溫裂解碳化后,制備了C3N4負載的單原子Ni-C-N催化劑。Deng等[55]以SiO2為模板,將(NH4)6Mo7O24與Co(NO3)2、CS2的混合物高溫裂解后,制備了單原子Co摻雜三維多孔結構的MoS2催化劑。
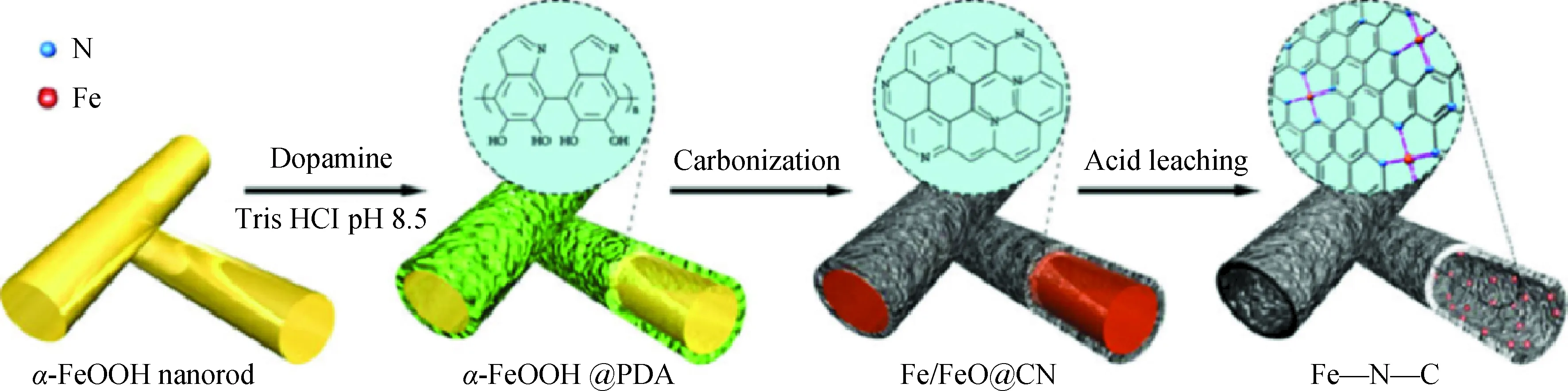
圖4 單原子Fe-N-C的制備示意圖[53]Fig.4 Schematic illustration of the synthesis of single atom Fe-N-C[53]
2 非貴金屬單原子催化劑的結構表征
理解單原子劑的結構及單原子催化的基本機理對于設計高性能和高穩定性的催化劑體系至關重要[56]。包括HAADF-STEM、XAFS、X-射線光電子能譜(XPS)、穆斯堡爾譜等在內的表征手段以及理論計算的發展對于單原子催化劑的發現、開發、優化及其催化機理的理解等方面具有不可替代的作用。其中,HAADF-STEM是唯一能夠直接“看到”催化劑中單個原子的工具,可提供具有空間分辨的局部信息,對于含有從微米到納米不同級別不均一性的多相催化劑而言,該技術尤其具有價值[57];包括X射線吸收近邊結構(XANES)以及擴展X射線吸收精細結構(EXAFS)的XAFS則能夠強有力地解析中心吸收原子的配位環境及電子態信息[58];XPS可以對中心金屬的電子態信息及部分配位環境進行輔助驗證;穆斯堡爾譜則可以分析Fe基單原子催化劑中Fe的超精細結構。只有將這些表征方法及其他分析手段合理地組合并與理論計算相結合,才能對非貴金屬單原子催化劑的結構進行有效地表征。
如前所述,Liu等[39]以Mg(OH)2為模板通過高溫裂解法制備了Co-N-C單原子催化劑,并對其結構進行了解析。圖5(a)為Co-N-C的HETEM圖,可以看出無明顯的Co納米顆粒,說明Co可能以原子簇或單原子的形式存在。圖5(b)為HAADF-STEM圖,直接證明了Co以單原子的形式高密度地分散在碳載體上。圖5(c)為Co-N-C及對比樣品的XANES譜圖,Co-N-C在7714~7716 eV的邊前區內沒有吸收峰,說明Co-N-C中的CoN4位點不是平面結構。進一步利用密度泛函理論(DFT)對不同構型的CoN4的XANES譜圖進行計算,從圖5(d)可以看出,CoN4C8-1-2O2構型的計算值與實驗表征結果最為一致。此時,Co中心與4個吡啶N成鍵,形成變形的平面正方形配位結構;軸向則有2個弱吸附的O2分子吸附在Co原子上。圖5(e)與(f)為經過傅里葉變換的EXAFS譜圖,通過與Co箔以及Co3O4的對比,可以看出Co-N-C中無Co-Co鍵及Co-O鍵,再次證明了Co的單原子分布;并且EXAFS譜圖的擬合結果與實驗表征結果具有一致性,驗證了模型的準確性。
Liu等[40]還對以納米MgO為模版,不同溫度下高溫裂解制備的單原子Fe-N-C催化劑的活性物種進行了結構解析。HAADF-STEM、EXAFS分析表明,600℃、700℃制備的Fe-N-C-600以及Fe-N-C-700中Fe物種原子分散,Fe-N-C-800中則存在少量的Fe納米顆粒。對應的XANES譜圖在7717 eV的邊前區內有明顯吸收峰,表明3種催化劑中FeN4為平面正方形配位結構。此外,XPS給出Fe的價態為Fe3+。圖6為Fe-N-C-600/700/800的57Fe穆斯堡爾譜圖[40]。從圖6可以看出,Fe-N-C-600/700的譜圖可由3條雙峰擬合,而Fe-N-C-800的譜圖由2條雙峰、1條單峰、1條線峰擬合。其中,單峰與六線峰表明Fe-N-C-800中存在γ-Fe以及FexC物種。進一步根據同質異能移、四級分裂值對雙峰進行分類,將上述雙峰分為D1、D2、D3、D4共4種類型。D1歸屬于類卟啉鐵FeⅡN4物種,D2為高自旋態的X-FeⅢN4-Y物種(X、Y為配位的O或N),D3為低自旋態的N-FeⅢN4-N 物種,D4則為5配位中自旋態的N-(FeⅢN4) 物種。這些結果表明,即使是單原子分散的Fe-N-C,Fe仍以多種形態分布,而有效地表征這些物種,是進一步研究其構效關系的基礎。
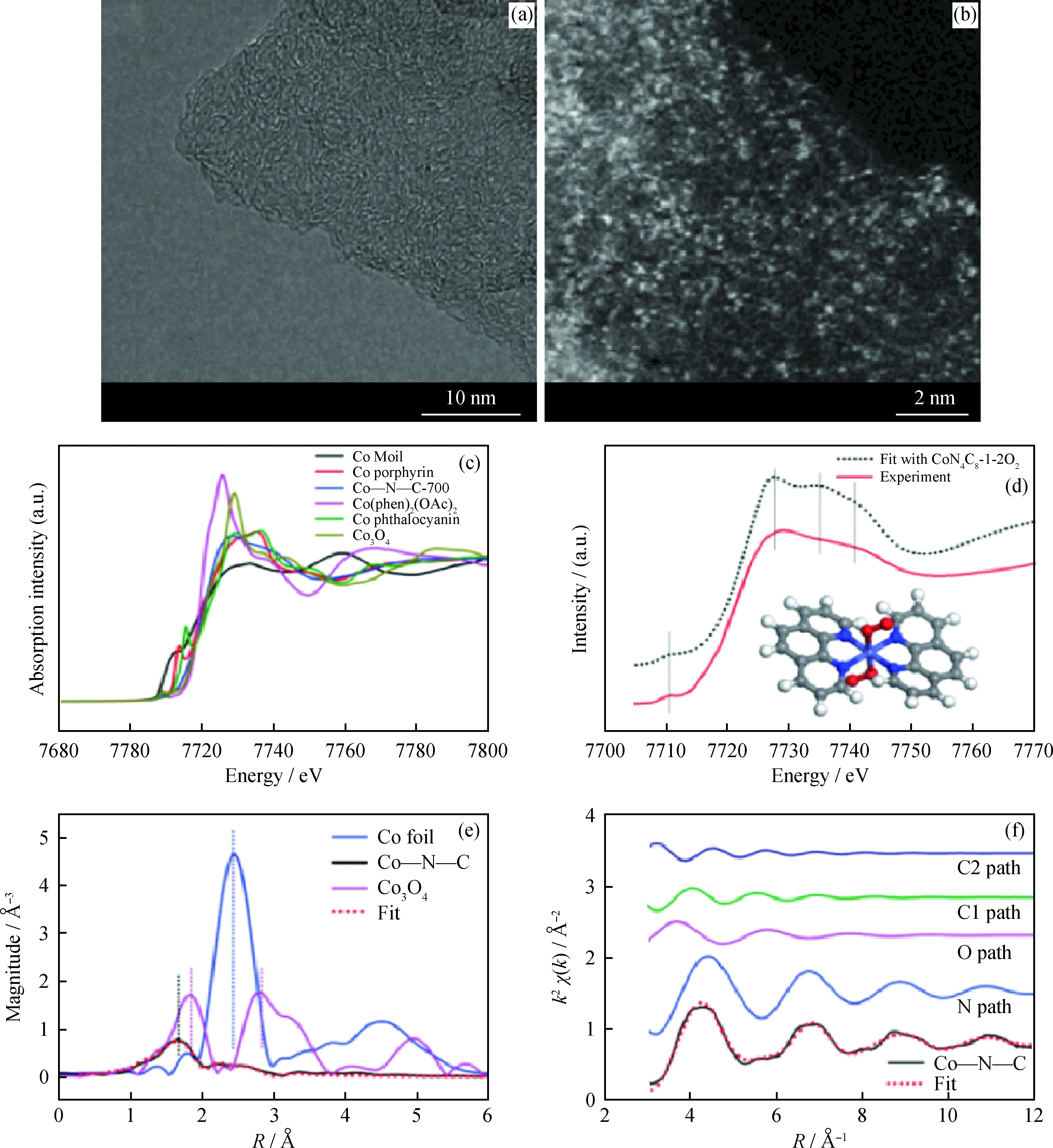
圖5 單原子Co-N-C催化劑的結構解析[39]Fig.5 Structure analysis of single-atom dispersed Co-N-C catalyst[39](a) HRTEM and (b) HAADF-STEM images of Co-N-C catalyst; (c) The normalized XANES spectra at the Co K-edge of different samples and (d) Comparison between Co-N-C catalyst and the theoretical spectrum (black dotted line); (e) The k2-weighted Fourier transform spectra of different samples; (f) The contributions of different paths including Co-N (blue line), Co-O (pink line) and Co-C (green and navy bluelines) in k-space for the Co-N-C sample
六
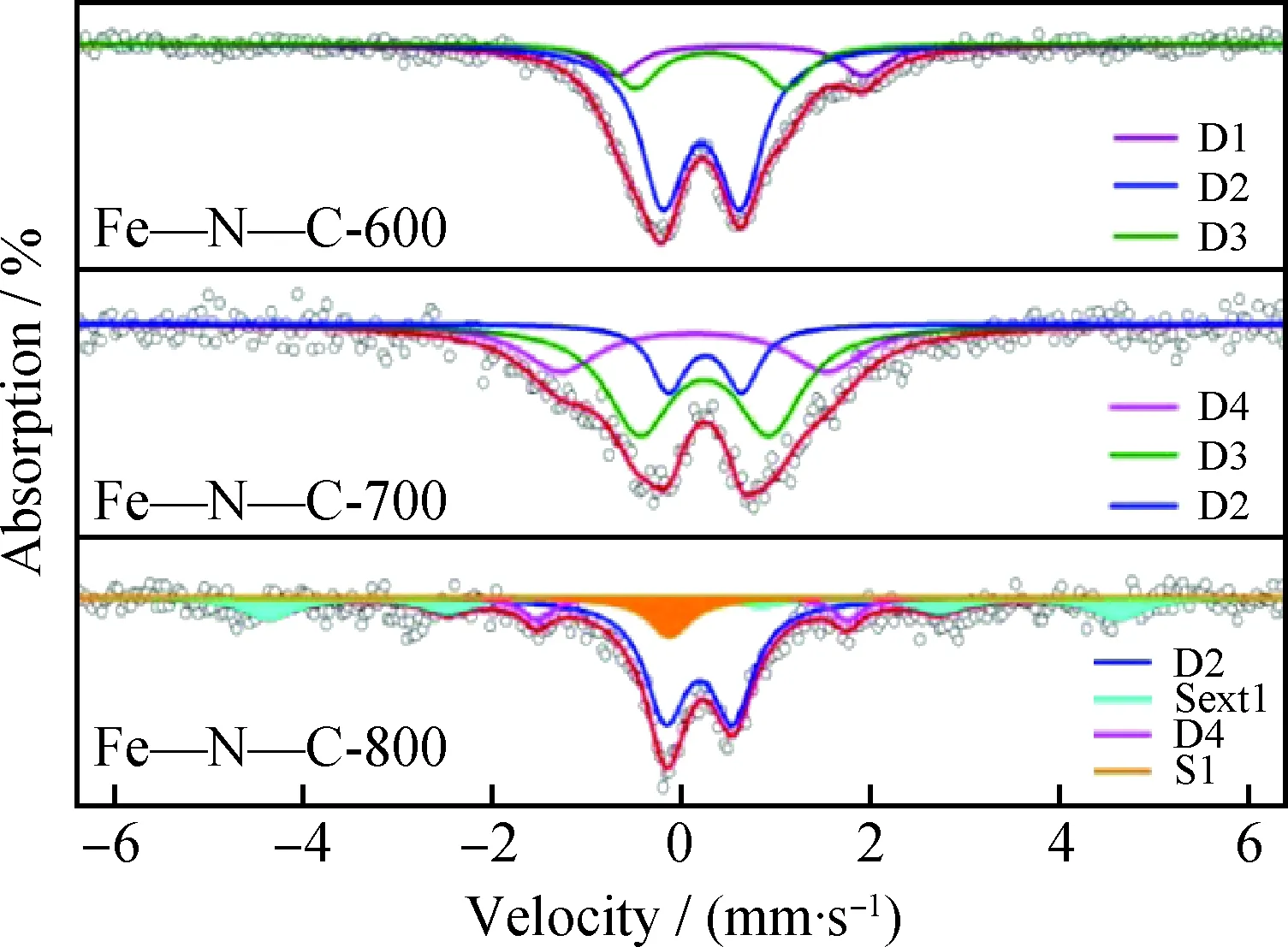
圖6 Fe-N-C-600/700/800的57Fe穆斯堡爾譜圖[40]Fig.6 57Fe M?ssbauer spectra of the Fe-N-C-600/700/800[40]
3 非貴金屬單原子催化劑的應用
3.1 電催化反應
氧還原反應(Oxygen reduction reaction, ORR)、析氧反應(Oxygen evolution reaction, OER)和HER是電解水、燃料電池、金屬-空氣電池等綠色能源技術的核心,催化劑對于促進這些反應起到重要作用[59]。目前常用的ORR和HER的催化劑是Pt基材料,而OER的催化劑則是IrOx或RuOx。基于導電多孔碳載體構建的單原子Co(Fe)-N-C 催化劑,由于具有良好的耐酸堿、抗毒化、離子、電子傳輸性能,并且利于氣相反應物、產物的擴散,被視為一種潛在的、可替代貴金屬催化劑的高性能電催化材料。
Liang等[37]將裂解法制備的Co(Fe)-N-C應用于酸性條件下的ORR。其中以SiO2納米顆粒為模板制備的Co-N-C具有較高的比表面積、均一的孔分布以及分散的Co-N-C位點,表現出最好的催化性能,ORR半波電位0.79 V,僅比20%Pt/C負移58 mV,且經循環伏安10000圈加速老化測試(AAT)后半波電位僅負移9 mV。Zhang等[52]以粒徑50 nm的FeZnZIF為前驅體制備的Fe-N-C催化劑,在酸性條件下的ORR半波電位達到了0.85 V,略低于Pt/C(60 μg Pt/cm2)的0.88 V,AAT實驗表明,催化劑的穩定性遠優于Pt/C;此外催化劑在ORR過程中H2O2的產率小于1%,表明此反應是1個4電子還原過程。最近,Shen等[38]的研究表明,在Fe-N-C中摻入S形成C-S-C 位點,有利于提升催化劑在酸性條件下的ORR性能。密度泛函理論(DFT)計算結果表明,當S與Fe的距離大于0.73 nm時可有效減少Fe中心的電子局域,促進Fe費米能級附近電子態與ORR中間態氧物種的相互作用,從而有效降低過電位。而在堿性條件下,Yin等[49]制備的單原子Co-N-C催化劑ORR半波電位0.881 V,優于20%Pt/C的0.811 V。Chen等[51]制備的單原子Fe-N-C催化劑ORR半波電位更是達到了0.900 V,并且具有傳統Pt/C催化劑所不具備的優異的抗甲醇毒化能力以及穩定性。DFT計算結果表明,單原子分散的FeN4中心有利于電子傳輸以及中間產物OH吸附物種轉化為OH-,是催化劑具有優異性能的關鍵。
在OER的應用研究方面,Li等[60]的理論計算結果表明,石墨相氮化碳(g-CN)負載的單原子Co、Ni催化劑具有較好的應該前景。Ohn等[54]則制備了g-CN負載的單原子Ni并應用于OER,結果表明,負載單原子Ni的g-CN的OER性能有顯著提升,但其催化過電位與IrOx催化劑仍有一定差距。Zheng等[45]的DFT計算結果表明,C3N4空穴穩定的單原子Co所形成的CoN3C2位點具有與貴金屬催化劑相近的OER及ORR催化性能。基于此結果,他們制備了包覆C3N4的CNT負載的單原子Co催化劑,并在1 mol/L的KOH電解液中評價OER性能,在電流密度達到10 mA/cm2時,對應的電位1.61 V,略高于IrO2催化劑。而經過900℃高溫處理破壞C3N4結構的催化劑性能明顯下降,表明C3N4與Co形成的CoN3C2是實際的活性位點。此外,Chen等[44]制備的CNT負載S摻雜的單原子Fe-N-C同樣是OER、ORR雙功能催化劑,并且以此催化劑構建的鋅-空氣電池器件在功率密度以及循環性能等方面均優于對應的20% Pt/C催化劑。通過與不含S的催化劑比較,他們認為,S、N對于C骨架的電荷分布以及活性位點FeNx的3d電子密度的影響是提高催化性能的關鍵。
在HER的應用研究方面,Qiu等[22]制備的單原子Ni催化劑在酸性條件下表現出突出的催化性能,起始過電位約為50 mV,Tafel斜率為45 mV/dec。XPS與理論計算表明,Ni嵌入到石墨烯層內,與C之間鍵和并發生電子轉移,形成空的雜化軌道是單原子Ni催化劑具有突出HER催化活性與穩定性的關鍵。Fan等以原位活化法制備的石墨烯負載單原子Ni催化劑同樣具有優異的HER催化性能,Tafel斜率為41 mV/dec,在電流密度為100 mA/cm2時,過電位僅為112 mV。此外,Fei等[43]制備的N摻雜石墨烯負載的單原子Co催化劑,在酸性條件下HER起始過電位為30 mV,Tafel斜率為82 mV/dec。通過比較不同Co負載量及N摻雜量的催化劑,他們認為,活性位點是與N配位的Co中心。總體而言,對于ORR、OER以及HER,非貴金屬單原子催化劑相較于貴金屬催化劑在抗毒化以及穩定性方面有明顯的優勢。尤其在堿性條件下的ORR,非貴金屬單原子催化劑的催化活性甚至優于貴金屬催化劑;但對于OER以及HER,其催化活性仍需進一步提高。
3.2 加氫及脫氫反應
偶氮苯類化合物是一種廣泛使用的染料及醫藥中間體,通常由貴金屬催化劑催化加氫芳香硝基化合物制得[61]。Liu等[39]以高溫裂解法制備單原子Co-N-C催化劑,并首次將非貴金屬單原子催化劑應用于此類加氫反應。Co-N-C催化劑在3 MPa H2、80℃的反應條件下,即可將硝基苯轉化為偶氮苯。該催化劑具有優異的底物普適性,即使苯環側鏈含有-C=C,-I,-Br等可還原性基團時,也可制備相應偶氮苯衍生物,催化選擇性明顯優于傳統的貴金屬催化劑。傅里葉變換衰減全反射紅外光譜表明,反應物的-C=C等官能團在Co-N-C 活性中心沒有發生吸附,是其具有高選擇性的主要原因。Zhu等[33]發現,單原子Cu催化劑可應用于5-羥甲基糠醛低溫加氫制2,5-呋喃二甲醇,催化活性比傳統沉淀法、浸漬法制備的含Cu納米顆粒催化劑的活性高了1、2個數量級。進一步分析表明,單原子Cu在載體表面形成Cu0-Cu+協同位點。其中,Cu0主要起解離H2的作用,Cu+作為親電位點則有利于反應物中C=O的吸附與活化。Li等[20]將NU-1000負載的單位點Ni催化劑應用于乙烯的加氫反應,其在50℃下單一Ni催化乙烯加氫的轉化速率(TOF)約為0.9 s-1,并且催化劑在100℃反應14 d后仍保留90%的初始活性。此單位點Ni催化劑在氯化二乙基鋁存在的條件下,還可有效的催化乙烯聚合,其TOF約為0.07 s-1,優于其他基于MOF構建的烯烴聚合催化劑。最近Dai等[50]則將單原子Ni-N-C催化劑應用于乙炔選擇性加氫制乙烯,在乙炔高轉化率(>90%)的條件下,乙烯的選擇性仍接近90%,這一性能甚至優于貴金屬Au-Pd催化劑。進一步分析表明,NiN4位點中N對單原子Ni的d電子軌道的調控,是催化劑取得高選擇性的關鍵。
在脫氫反應的應用研究方面,Guo等[34]將Fe?SiO2應用于甲烷制乙烯、芳烴的反應中。研究表明,甲烷分子在配位不飽和的Fe中心上催化活化脫氫,獲得表面吸附態的甲基物種。在傳統催化劑表面,吸附態甲基物種容易進一步脫氫或者發生C-C偶聯而造成積炭失活。而單位點分散的Fe由于缺少相鄰的Fe-Fe位點,甲基物種容易從表面脫附形成高活性的甲基自由基,隨后在氣相中經自由基偶聯反應生成乙烯和其它高碳芳烴分子。如圖7所示,甲基物種在單位點Fe上脫附形成甲基自由基的能壘僅有2.32 eV;Fe中心吸附第2個甲烷分子并形成甲基自由基的能壘則分別為3.07和2.19 eV。在反應溫度1090℃和空速21.4 L/(gcat·h)條件下,單位點Fe催化劑的甲烷單程轉化率達48.1%,乙烯的選擇性為48.4%,所有產物(乙烯、苯和萘)的選擇性>99%,實現了CO2的零排放。此外,Li等[21]將NU-1000負載的單位點Co催化劑應用于丙烷的氧化脫氫,催化劑在反應溫度200℃下即表現出了明顯的催化性能。然而當丙烷轉化率超過10%時,大部分丙烷發生深度氧化,丙烯的選擇性不足30%,如何實現低溫下選擇性氧化脫氫仍是該研究領域的難點。
3.3 液相氧化反應
苯酚是重要的有機化工原料,溫和條件下、高效的將苯直接羥化合成苯酚具有重要的經濟意義與研究價值,是催化領域的一個研究熱點[62]。Deng等[27]研究發現,FeN4/GN即使在0℃的條件下,以H2O2為氧化劑即可催化氧化苯制苯酚。而在25℃反應溫度下,FeN4/GN催化苯轉化的初始TOF為84.7 h-1,反應24 h后苯的轉化率為23.4%,苯酚的產率為18.7%。利用DFT計算,他們認為,FeN4的中心Fe原子通過解離H2O2形成O=Fe=O,這一位點可以有效地吸附苯分子,隨后發生氫轉移反應將苯直接氧化成苯酚。XAFS表征結果也支持催化劑活性中心O=Fe=O的形成。Zhang等[53]同樣將單原子的Fe-N-C應用于催化氧化苯制苯酚。他們認為,Fe首先將H2O2分解形成2個羥基自由基,隨后生成1個水分子以及活性氧物種,活性氧物種直接將苯氧化成苯酚。除了苯的催化氧化外,Liu等[40]以叔丁基過氧化氫為氧化劑,將Fe-N-C應用于乙苯的選擇性催化氧化,苯乙酮的選擇性高達99%。并且該催化劑具有優異的底物普適性,即使苯環側鏈含有-OCH3等給電子基團或者-NO2等吸電子基團,或者反應物為雜環化合物以及環己烷時,也可高選擇性地制備相應酮類化合物。為了探究反應的活性位點,以KSCN為滴定劑對Fe中心進行選擇性毒化并比較催化性能,結合穆斯堡爾譜的表征,推斷中自旋態的N-(FeⅢN4)是活性最高的FeN4物種。此外,Zhang等[42]以Co-N-C為催化劑,O2為氧化劑,催化氧化一級醇與二級醇脫氫偶聯,制備α,β不飽和酮。Xie等[63]以Cu(Fe, Co, Ni, Cr)-N-C 為催化劑,O2為氧化劑,催化氧化醇,制備對應的醛。
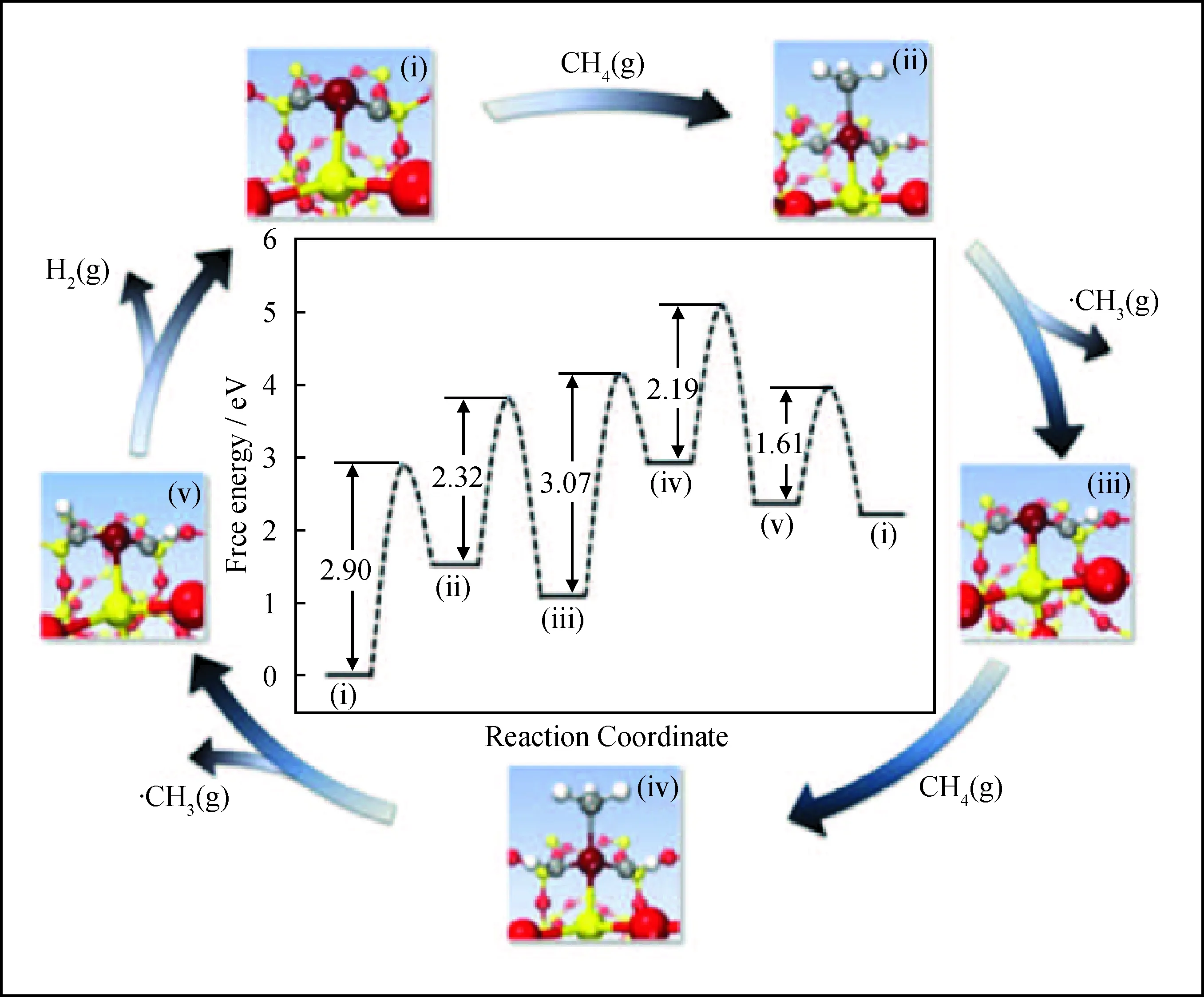
圖7 Fe?SiO2在1223 K催化甲烷形成甲基自由基的能量剖面圖[34]Fig.7 DFT calculations on catalytic generation of methyl radicals over Fe?SiO2 at 1223 K[34]
3.4 光催化還原CO2及其他反應
CO2的資源化再生利用,對于同時解決溫室效應與能源短缺兩大問題具有重要的理論價值和現實意義,其中光催化還原CO2是目前最主要的研究方向之一[64]。Zhang等[23]比較了MOF-525負載了單位點Co前后的可見光催化還原CO2性能。其中MOF-525-Co在以三乙醇胺為電子犧牲體的條件下,光催化CO2生成CO的速率為200.6 μmol/(g·h),生成CH4的速率則為36.67 μmol/(g·h)。研究表明,MOF-525的卟啉環引入單原子Co后,極大的提升了卟啉環的電子空穴分離效率,延長了光生電子的壽命并促進了吸附在Co中心的CO2的還原。Long等[30]制備的Pd7Cu1-TiO2光催化還原CO2生成CH4的速率為19.6 μmol/(g·h),選擇性達到96%。理論計算與實驗結果共同表明,單原子分散的Cu起到了2個關鍵作用:(1)形成配對的Pd-Cu位點促進了反應物CO2的吸附,并且抑制了副產物H2的生成;(2)Pd晶格調控了Cu位點的d帶中心位置,有利于吸附的CO2的活化。除了光催化還原CO2外,Liu等[65]近期的研究表明,單位點的Co催化劑可以有效分離和傳輸光生電子和空穴,實現了高效、自發的太陽光催化全解水析氫。此外,近期的研究結果還表明,非貴金屬單原子催化劑可應用于染料敏化太陽能電池[28],電催化還原CO2[66],CO的催化氧化[67-69],水煤氣變換[70]等一系列重要反應。
4 結論與展望
單原子催化這一多相催化領域的新概念的提出,在一定程度上模糊了多相催化與均相催化的界限,將金屬催化劑的原子利用率提高到新的高度,促進對多相催化過程與機理的認識向原子級別層面的深入。從金屬催化劑自身成本的角度出發,非貴金屬催化劑相對于貴金屬催化劑具有天然的優勢;自然界中存在的幾類十分重要的高效非貴金屬單原子催化劑則為非貴金屬單原子催化劑的發展樹立了標桿與方向。
當前,非貴金屬單原子催化劑的制備與應用研究方面已取得了諸多進展,但也存在著許多難題與挑戰:(1)無論是基于特定設備的ALD法、電弧放電法,或是以金屬配位有機化合物為前驅體的高溫裂解法、球磨法等制備的非貴金屬單原子催化劑成本仍相對較高,并且在催化劑的可控制備、宏量制備方面還存在著許多不足;(2)載體的類型與結構對于單原子催化劑的性能起著重要作用,目前已成功研究的載體僅限于碳載體、硅載體、MOF等少數類型,如何擴展載體類型以提升催化性能甚至構建雙功能催化劑仍有許多難點;(3)擴展Fe、Co、Ni外的非貴金屬單原子的制備與應用并提升非貴金屬的負載量;(4)對非金屬單原子催化劑的結構解析并闡明其在特定反應中的構效關系。
非貴金屬單原子催化劑的出現與發展為貴金屬催化劑的替代提供了新的可能,盡管該領域目前的研究仍處于實驗室階段且存在諸多挑戰,但其迅猛的發展勢頭與廣闊的發展前景勢必將推動該領域的研究不斷向工業化應用方向前行。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
