小分子海洋抗菌肽鱟素Ⅰ里氏木霉表達系統構建
2018-08-08 08:23:34張麗君代建國
江蘇農業科學 2018年14期
張麗君,王 英,張 燕,簡 琛,金 剛,代建國
(1.深圳職業技術學院應用化學與生物技術學院,廣東深圳 518055; 2.廣州城市職業學院食品系,廣東廣州 510405)
陽離子抗菌肽(antimicrobial peptide,AMP)鱟素Ⅰ(tachyplesin Ⅰ)由Nakamuro等首次從中國鱟(Tachypleustridentatus)血細胞分離純化制得[1],分子量為2 263 u,一級結構由17個氨基酸殘基構成,含有2個二硫鍵;二級結構呈典型的反平行β-折疊結構,疏水的氨基酸殘基定位于平面一側,分子中6個陽離子氨基酸殘基主要分布在分子的尾部。鱟素Ⅰ具有抗細菌[2]、抗真菌[3]、抗病毒[4]、抑制腫瘤細胞增殖和誘導癌細胞分化[5]以及顯著調控動物腸道微生物菌群[6]的功能,極具潛在廣泛的應用價值。然而,天然鱟素Ⅰ來源非常受限,人工合成價格非常昂貴,因此基因重組制備成為規模化獲取鱟素Ⅰ的重要途徑。
鱟素Ⅰ對常見基因工程宿主的廣泛和強烈的抑殺活性[7-8]使其高效表達異常艱難,融合表達雖可降低鱟素Ⅰ對工程宿主的抑殺活性,但因特異性蛋白酶價格昂貴和后續切割精度難以保證而無法得到有效應用。因此,從天然微生物篩選得到耐受鱟素Ⅰ抑殺的宿主以提高鱟素Ⅰ的表達效率值得期待。本課題組比較了鱟素Ⅰ對大腸桿菌(BL21)、枯草芽孢桿菌(WB800、BS168)、酵母菌(GS115)、小球藻和絲狀真菌里氏木霉(QM9414)等常見工程宿主的抑殺活性,發現絲狀真菌里氏木霉對鱟素Ⅰ的耐受能力最強[7-8]。迄今為止,抗菌肽能否在里氏木霉中獲得有效表達尚未見相關報道。本研究擬構建鱟素Ⅰ-里氏木霉表達系統,探索鱟素Ⅰ在此表達系統中的表達效率。
1 材料與方法
1.1 材料
1.1.1 試驗材料 里氏木霉表達載體pAN-PSGT和里氏木霉(QM9414),均由深圳大學劉剛教授惠贈;大腸桿菌(Escherichiacoli) Competengt Cell JM109,購自大連TaKaRa公司。
1.1.2 試劑 質粒快速提取試劑盒、瓊脂糖凝膠DNA純化回收試劑盒、DNA連接試劑盒、Trizol試劑盒和兩步法RT-PCR試劑盒以及蛋白質Marker,均購自TaKaRa公司;溶壁酶、氨芐青霉素和潮霉素,均購自Sigma公司;NotⅠ、SfiⅠ酶和X-gal,均購自TaKaRa公司。
1.2 方法
1.2.1 天然鱟素基因(Tac)序列的人工合成 從GenBank中獲得鱟素Ⅰ的堿基序列,根據絲狀真菌里氏木霉偏愛密碼子優化其序列:在其5′端和3′端分別加上NotⅠ和SfiⅠ酶酶切位點,并補齊宿主中信號肽的部分序列,設計目的基因序列如下:
5′-GGCCTTCTTGGCCACAGCTCGTGCTAAGTGGTGCTT-CCGCGTCTGCTACCGCGGCATCTGCTACAGGCGATGTCGCTA-AGCGGCCGC-3′,該序列全長87 bp,委托大連寶生物公司人工合成。
1.2.2 鱟素-里氏木霉表達載體構建 pAN-PSGT全序列9 710 bp,包括Pgpd啟動子序列(2 129 bp)、潮霉素抗性基因php序列(1 023 bp)、終止子序列TtrpC(770 bp)、PUC18中從SALI到ECORI之間的序列AP(2 648 bp)、gpd啟動子序列(1 347 bp)、信號肽序列CBHⅠ sig(50 bp)、LacZ序列(1 132 bp)、Tcbh終止子(340 bp)。采用常規分子重組技術將鱟素Ⅰ基因Tac連接到gpd-sig和Tcbh中間,構建成一個新的表達載體pAN-PSGT-Tac,轉化大腸桿菌JM109,采用α互補法(藍/白斑篩選法)進行陽性克隆篩選。菌落PCR法鑒定轉化子,用通用引物M13-47/RV-M進行PCR擴增。反應體系總體積50 μL,PCR反應條件為:94 ℃ 1 min;94 ℃ 30 s,55 ℃ 30 s,72 ℃ 1 min,30個循環;72 ℃ 7 min。用2%瓊脂糖凝膠電泳檢測PCR產物。PCR產物進一步經大連寶生物有限公司測定序列。
1.2.3 重組載體pAN-PSGT-Tac轉化里氏木霉原生質體 里氏木霉原生質體的制備和轉化在參考Penttila等的方法[9]基礎上改造如下:里氏木霉孢子接種于PDA固體培養基,28 ℃ 培養7 d;無菌水制備孢子懸液,取0.8×108~ 1.0×108個孢子,接種到40 mL液體培養基中,于28 ℃、250 r/min 培養11~15 h;待孢子萌發,取25 mL菌液置于 25 mL 離心管,6 000 r/min 離心5 min;去上清,菌絲用1 mol/L MgSO4洗2遍,8 000 r/min 離心5 min;用10 mL酶解液(100 mg 溶壁酶溶于 10 mL 1 mol/L MgSO4后過濾除菌)重懸菌絲并置于100 mL三角瓶中,28 ℃、70 r/min培養2.0~2.5 h,鏡檢原生質體的情況。在酶解液中加入1~2倍體積的STC[1.2 mol/L山梨醇,10 mmol/L Tris·Cl(pH值為7.5),50 mmol/L CaCl2]8 000 r/min 離心15 min,沉淀原生質體。去上清,用STC溶液洗滌原生質體2次,8 000 r/min離心10~15 min,最后將原生質體懸浮于1 mL STC溶液中。調整原生質體的濃度為108個/mL,取200 μL,加入20 μg pAN-PSGT-Tac(總體積不超過20 μL),輕輕混勻;48 ℃熱激2 min;加入50 μL 60%的PEG-4000(用pH值為7.5含50 mmol/L CaCl2和 10 mmol/L Tris·Cl的緩沖液配制),于室溫靜置20 min。溶液先轉移到25 mL離心管中,再加入 2 mL 60%的PEG-4000,混勻后于室溫靜置5 min;加入 20 mL STC,11 000 r/min,離心15 min;用1 mL STC溶液重懸原生質體沉淀后加入到10 mL原生質體液體再生培養基中,于28 ℃、70 r/min條件下培養20~24 h(原生質體長成微小菌絲即可,若菌絲較大,則涂板后不易區分單菌落);8 000 r/min 離心10 min后用0.5~1.0 mL STC溶液懸浮沉淀,將懸浮液涂布于5個含 100 μg/mL 潮霉素B的PDA固體平板(不含STC)上,28 ℃培養2 d左右;待平板上長出菌絲(平板底部變黃),將菌絲連同其下方的一層薄薄的瓊脂塊一同挖下,菌絲朝下接到新的PDA固體小平板上(含80 μg/mL潮霉素B),28 ℃培養7 d左右。
1.2.4 里氏木霉轉化子鑒定
1.2.4.1 轉化子潮霉素基因鑒定 將篩選平板上長出的潮霉素抗性轉化子分別轉接到PDA平板上進行擴大培養,1周后首先將各轉化子分別接種于液體基本培養基中,提取各轉化子的基因組DNA作為模板,用引物HPH1和HPH2進行PCR擴增,擴增產物進行瓊脂糖凝膠電泳檢測[10]。
1.2.4.2 轉化子的基因型鑒定 提取轉化子基因組DNA作為模板,由于Tac基因太小,PCR擴增存在極大難度,本試驗設計的鑒定方式為擴增Tac基因上下游的片段,分別為gpd-sig-tac-Tcbh、gpd-sig、gpd-sig-tac、tac-Tcbh和Tcbh,如果能擴增出這5種片段,則可確定鱟素Ⅰ基因Tac已成功同源替換到里氏木霉基因組DNA上了,并且插入方向正確。擴增片段、所需引物和預計擴增片段大小見表1。并將用引物GPD1和TRD3擴增所得的PCR產物進行核苷酸序列測定。
1.2.5 引物設計與合成 本研究過程所采用的引物序列見表2,各引物均由大連寶生物公司合成。
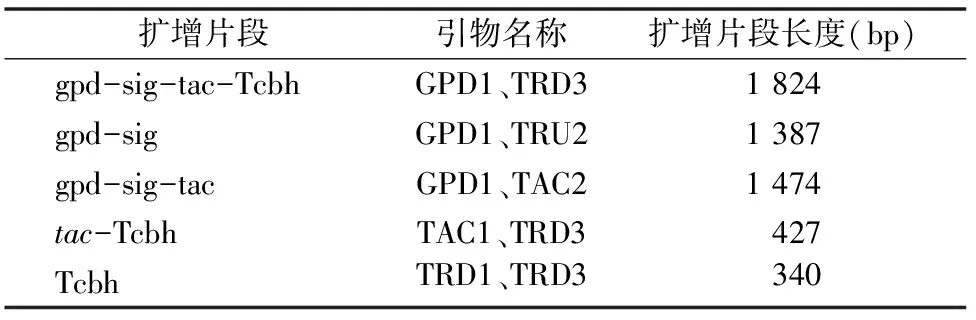
表1 擴增片段、所需引物和預計擴增片段大小

表2 引物序列
2 結果與分析
2.1 pAN-PSGT-Tac重組載體構建
以重組載體為模板,采用PAN-PSGT通用引物M13-47/RV-M進行PCR擴增。通用引物擴增產物大小約 150 bp,加上鱟素Ⅰ基因87 bp,預計目的條帶大小約為 240 bp,電泳結果見圖1,在250 bp附近發現1條清晰的條帶,初步確認其為目的條帶。PCR產物測序結果與已知序列同源性達到100%,確認鱟素Ⅰ基因Tac已經正確連接至表達載體PAN-PSGT之中。

2.2 載體pAN-PSGT-Tac對里氏木霉原生質體轉化后的鑒定
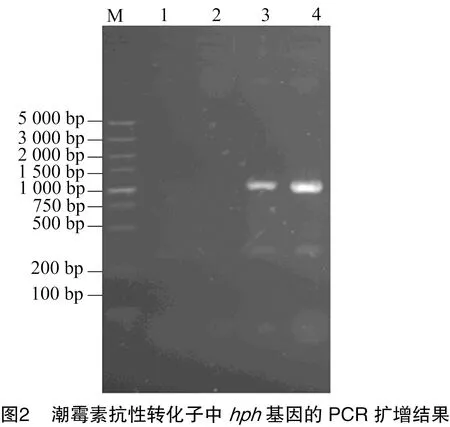
2.2.1 潮霉素基因的PCR擴增結果 提取篩選平板上長出的潮霉素抗性轉化子的基因組DNA為模板,PCR擴增潮霉素抗性基因,引物為HPH 1和HPH 2。由圖2可知,潮霉素抗性基因hph基因大小為1 023 bp,在1 000 bp處發現條帶,可以證明潮霉素基因成功擴增,初步確定潮霉素基因已經成功轉化。
2.2.2 轉化子基因型鑒定 取潮霉素基因鑒定為陽性的菌株,提取其基因組DNA按“1.5.2”節進行基因型鑒定,由圖3可知,以基因組DNA為模板,1泳道為擴增出的gpd-sig-tac-Tcbh片段,擴增條帶在1 500~2 000 bp之間,符合預期大小1 824 bp;2泳道為擴增出的gpd-sig片段,大小在1 000~1 500 bp 之間,符合預期大小1 387 bp;3泳道為擴增出的gpd-sig-tac片段,大小在1 500 bp附近,符合預期大小 1 474 bp;4泳道為擴增出的tac-Tcbh片段,大小靠近 500 bp,符合預計大小427 bp;5泳道為擴增出的Tcbh片段,大小在250~500 bp之間,符合預計大小340 bp。5種擴增都驗證為正確,即可初步確定鱟素Tac基因已正確同源整合到基因組DNA上。針對gpd-sig-tac-Tcbh的擴增產物測序結果與已知序列的同源性達100%,可以證明鱟素基因已經重組到基因組DNA上。

3 討論
基因重組表達鱟素Ⅰ的研究開始于20世紀90年代,但重組鱟素Ⅰ商品至今尚未問世。總體而言,國內外對鱟素Ⅰ基因工程表達主要集中在以細菌、真菌和藻類為宿主的表達體系上。
張春義等首次在大腸桿菌中以包涵體形式表達了鱟素Ⅰ,但因需變性和復性,產品下游處理難度較大[11-12];Dai等在枯草桿菌WB800中分泌表達了鱟素Ⅰ,但表達量最高僅達到10 μg/mL,與酵母菌是否可分泌性表達鱟素Ⅰ報道結果[6]相差較大。張春義等報道鱟素基因可克隆至畢赤酵母表達載體,其誘導分泌產物有抑菌效果,但具體蛋白表達量均未提及[13-14];Xu等將鱟素Ⅱ克隆至畢赤酵母SMD1168,鱟素Ⅱ表達量經過甲醇誘導6 d可達150 mg/L[15];Dorrington未能在畢赤酵母發酵液中檢測到鱟素Ⅰ[16];Gao等實現了鱟素Ⅰ基因和人溶菌酶基因在畢赤酵母的融合表達,雖然避免了鱟素Ⅰ對基因工程宿主的抑殺作用,但用特異性蛋白酶切割鱟素Ⅰ與融合蛋白時,容易造成氨基端出現多余的氨基酸,或切割效率低下,甚至不能切割等[17];吳順章等[18]、鄧祥元等[19]分別成功地將鱟素Ⅰ基因整合到壇紫菜(紅藻門,多細胞真核藻)和海帶配子體(褐藻門,單細胞真核藻)內,但前一研究僅在DNA和mRNA層面檢測到鱟素Ⅰ基因整合到壇紫菜基因組中,未報告是否表達了鱟素Ⅰ,后一研究雖表達了具有活性的鱟素Ⅰ,但其表達量要低于大腸桿菌表達體系[11]和枯草桿菌表達體系[6,10,18-19]。
綜上所述,本課題組觀察了鱟素Ⅰ對大腸桿菌、枯草桿菌、酵母菌、小球藻和絲狀真菌里氏木霉的抑殺活性,發現除絲狀真菌里氏木霉外,鱟素Ⅰ對其余工程宿主的最小抑殺濃度均不超過20 μg/mL[7-8]。因此,本課題組猜想,鱟素Ⅰ可強烈抑殺工程宿主極有可能是其一直難以獲得高效表達的主要原因。因此,找到比較耐受鱟素Ⅰ抑殺的工程宿主,如絲狀真菌里氏木霉,可能是解決鱟素Ⅰ如何獲得高效表達的關鍵。
絲狀真菌里氏木霉生產纖維素酶、半纖維素酶和淀粉酶等已有多年的歷史,該菌株在產酶條件下不產生毒素。實踐還表明,經基因工程手段改造的里氏木霉表達系統能夠實現異源蛋白過量表達。Emilio等將TrichodermaharzianumP1的幾丁質酶編碼基因置于里氏木霉CBH Ⅰ啟動子之后,搖瓶產量可達130 mg/L(達到原產菌株的20倍)[20]。幾丁質酶能抑制多種真菌的生長,但里氏木酶卻能高效生產幾丁質酶。可在里氏木霉中獲得高效表達的異源蛋白還有肌醇六磷酸酶(2 g/L)、哺乳動物的牛凝乳蛋白酶(40 mg/L)、抗體片段(150 mg/L)和人N-乙酰葡萄糖苷轉移酶等[21]。但迄今為止,抗菌肽能否在絲狀真菌里氏木霉系統中得到高效表達尚不清楚。
本研究根據里氏木霉偏愛密碼子優化了鱟素Ⅰ的基因序列,人工合成了該序列,采用酶切、連接等方法將鱟素Ⅰ基因成功連接至里氏木霉表達載體pAN-PSGT之上,構建了 pAN-PSGT-Tac重組表達載體。pAN-PSGT表達載體無需“共轉法”,可“單轉法”即直接用質粒轉化里氏木霉原生質體;pAN-PSGT采用里氏木霉QM9414 CBH Ⅰ的信號肽來引導目的蛋白的分泌。Kuo等發現許多真菌的gpd啟動子可用于重組蛋白的調控和轉錄過程中,提高目的產物的表達水平[22]。同時,信號肽可引導目的蛋白分泌到胞外,產物分離時不必破碎宿主細胞,同時分泌到胞外的蛋白往往都具有正確的空間折疊構象,利于下游的分離純化;pAN-PSGT中帶有LacZ基因,可通過藍白斑篩選陽性克隆。
采用PEG介導法將重組表達載體pAN-PSGT-Tac轉化至里氏木霉QM9414原生質體,潮霉素篩選得到了重組子,基因型鑒定結果表明,鱟素Ⅰ基因已經重組到基因組DNA上,說明本研究成功構建了鱟素Ⅰ里氏木霉表達系統。利用本系統表達鱟素Ⅰ蛋白的研究需要進一步研究。
4 結論
本試驗成功構建了鱟素Ⅰ-里氏木霉表達載體,并成功轉化至里氏木霉QM9414細胞中。這一結果為進一步改進鱟素Ⅰ在里氏木霉表達系統中表達研究奠定了基礎,對于其他小分子肽類蛋白在絲狀真菌表達系統中的表達也具有一定的指導意義。
