SiO2氣凝膠微球及其性能研究*
2018-07-28 08:10:08劉霄昱陳琦峰黃智明曾令可
陶瓷 2018年6期
關鍵詞:實驗
劉霄昱 陳琦峰 黃智明 王 慧 曾令可
(華南理工大學材料科學與工程學院 廣州 510640)
前言
由于氣凝膠擁有大部分固體材料所不具有的獨特性能,因此在近幾十年來被廣泛研究。氣凝膠內部90%以上是由空氣組成,剩余的10%則是由固體部分組成。對SiO2氣凝膠而言,固體部分是由直徑為3~15 nm的SiO2顆粒所構成的三維網絡結構,其孔徑為30~50 nm[1],屬于介孔材料。通常氣凝膠的密度為0.12~0.3 g/cm3[2],比表面積為400~1 200 m2/g,熱導率低至0.06 W/(m·K)[3],孔隙率高達85%~99%[4]。正因為氣凝膠上述這些優異的性能,使其在熱學、聲學、催化方面有著廣泛的應用。如太陽能系統的超級絕熱材料[5],有效的催化劑和催化劑載體[6],火箭推進器中的儲液材料[7]等。SiO2氣凝膠的制備一般包含3個過程,即溶膠凝膠過程,凝膠的老化過程和凝膠的干燥過程。由于塊狀氣凝膠制備困難且強度低,如若使用常壓干燥方法,則需要進行長時間的溶劑交換和表面改性過程,使得制備需要耗時幾天甚至幾周的時間,大大制約了其工業化的進程。而溶劑交換的速度主要取決于溶劑的表面張力和凝膠的大小兩個方面,如果需要制備大的塊狀氣凝膠,則需要進行相當長時間的溶劑交換過程;如果能制備出顆粒較小的凝膠微球,那么可以使得溶劑交換的過程相較于制備大的塊狀氣凝膠來說更加地迅速,可降低生產的成本且縮短生產周期。本實驗使用廉價的硅溶膠為原料,以常壓干燥的方法來制備氣凝膠微球,一方面可避免制備塊狀氣凝膠的麻煩,另一方面凝膠微球狀的粉體在實際生產應用也比較廣泛。
1 實驗
1.1 實驗原料
實驗過程中的主要原料和試劑如表1所示。

表1 實驗原料和試劑Tab.1 Raw materials and reagents
1.2 實驗儀器及測試設備
實驗過程所用主要儀器及測試設備如表2所示。
1.3 實驗工藝路線
SiO2氣凝膠微球的制備流程如圖1所示。
表2實驗儀器及測試設備
Tab.2 Experimenting and testing & characterization equipments
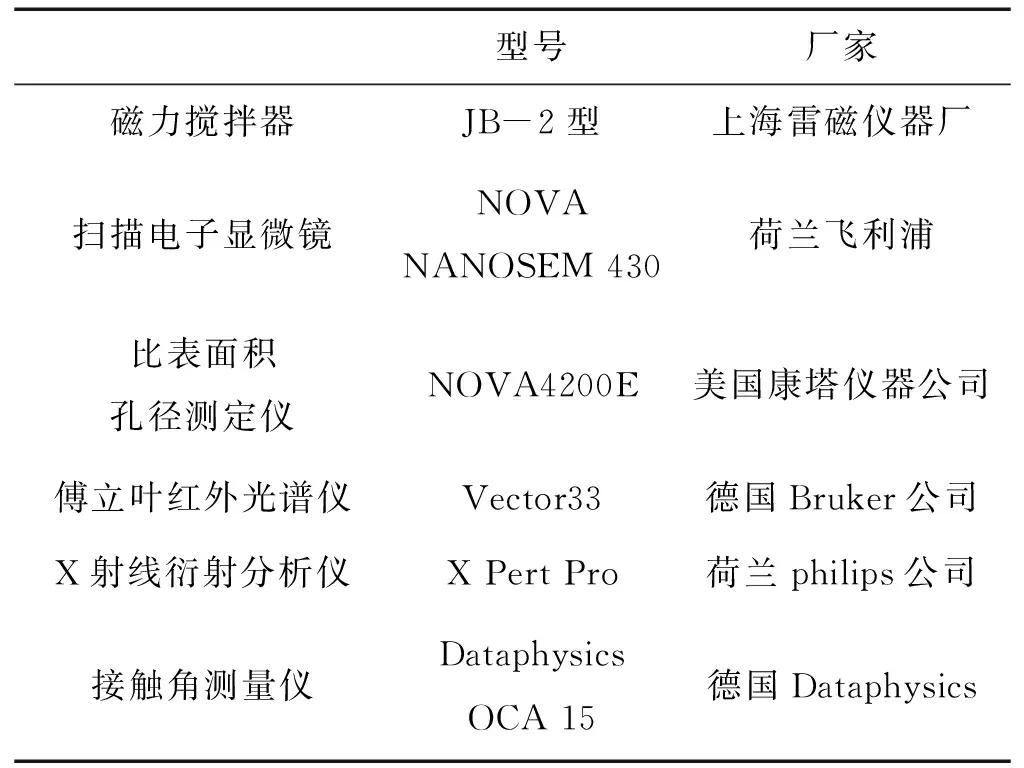
型號廠家磁力攪拌器JB-2型上海雷磁儀器廠掃描電子顯微鏡NOVA NANOSEM 430荷蘭飛利浦比表面積孔徑測定儀NOVA4200E美國康塔儀器公司傅立葉紅外光譜儀Vector33德國Bruker公司X射線衍射分析儀X Pert Pro荷蘭philips公司接觸角測量儀Dataphysics OCA 15德國Dataphysics
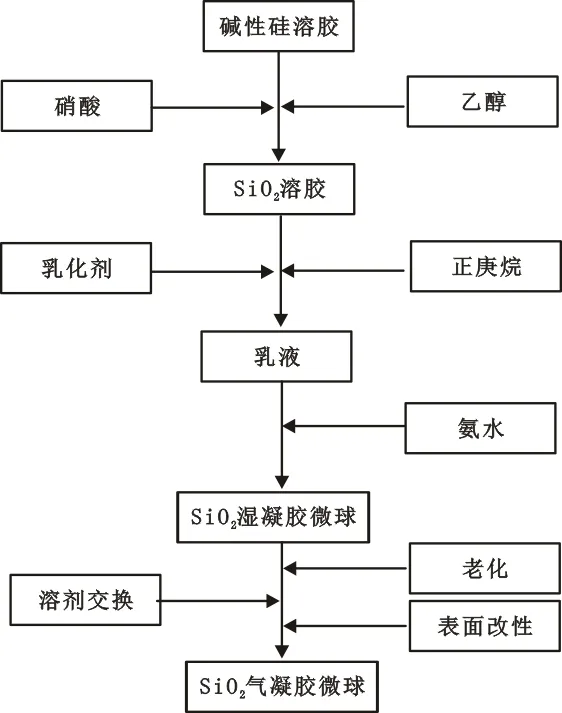
圖1 SiO2氣凝膠微球制備工藝流程圖Fig.1 Systematic preparation of silica aerogel microspheres
2 SiO2氣凝膠微球結構及性能研究
2.1 SiO2氣凝膠微球的形成機理
當硅溶膠在攪拌條件下均勻分散在油相中時,會在油相中形成微小的液滴,并且液滴的表面被一層表面活性劑所包圍以防止其相互聚集。圖2為SiO2氣凝膠微球的形成機理。為了簡潔易懂圖2中只標出了一個液滴。
凝膠微球的形成過程一般包括以下幾個步驟:
1)硅溶膠加入油相中形成小液滴。
2)在含有硅溶膠的油相中滴加氨水,氨水被分散成液滴。
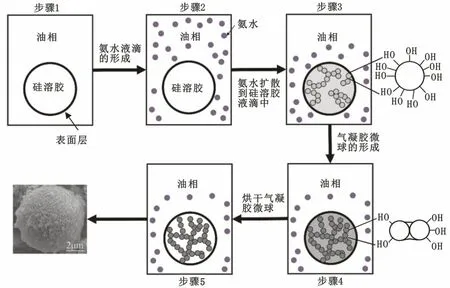
圖2 SiO2氣凝膠微球的形成機理Fig.2 Proposed mechanism for the formation of silica aerogel microspheres
3)氨水包圍在硅溶膠液滴表面并且在其表面發生聚合反應。
4)氨水擴散進入硅溶膠液滴內部并且在其內部均勻分散,使硅溶膠內部發生聚合反應,即濕凝膠的形成過程。
5)SiO2濕凝膠微球的干燥,去除凝膠內部孔隙中的水分。
雙表面活性劑在形成介孔凝膠微球的時候同樣也起到了重要的作用,因為這不僅可使液滴穩定防止其聚集成塊,同時也使得催化劑均勻地分散在液滴周圍使硅溶膠發生聚合反應。
2.2 工藝條件對SiO2氣凝膠微球形貌的影響
在本實驗中,采用乳液成球原理,可以通過控制液滴的尺寸來控制凝膠顆粒的大小。為了研究實驗過程中的參數對凝膠微球的大小尺寸的影響,考慮了復合乳化劑的配比、攪拌速度等因素。因此,筆者就對影響SiO2氣凝膠微球形貌的因素進行探索研究。表3為不同反應條件下所制備樣品。
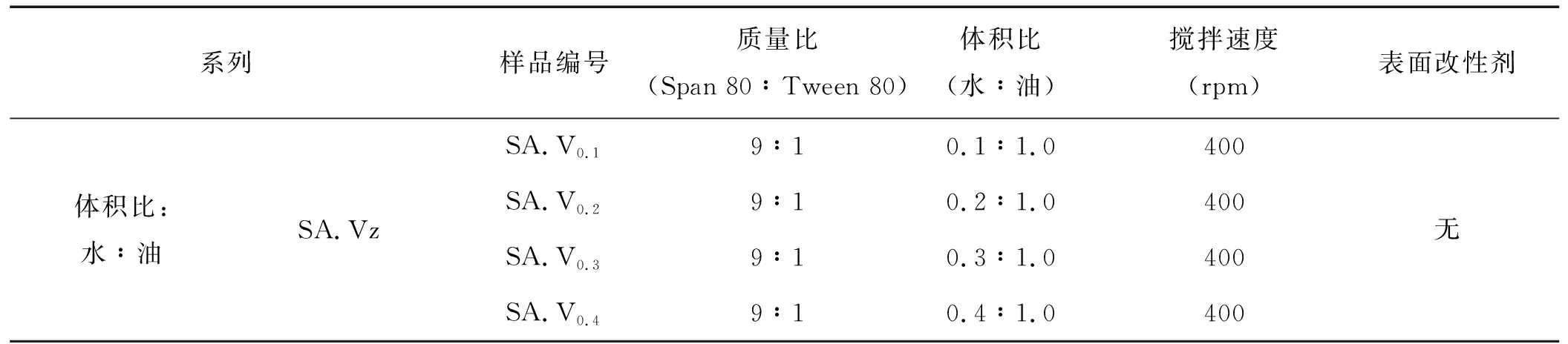
表3 不同反應條件下的樣品Tab.3 Identification of the samples prepared under various conditions

續表3
2.2.1 乳化劑配比對SiO2氣凝膠微球形貌的影響
圖3是乳化劑配比對凝膠微球粒徑的影響。
通過表5,可以看出學生在自主學習方面實驗班明顯好于對照班,這說明合作學習法可以提高學生參與課堂的積極性,學生可以自主選擇適合自己的教學方法,實現了因材施教和以學生為主體的教學目標。

(a, d) SA. S8∶T2 (b, e) SA. S9∶T1和 (c, f) SA. S9.8 ∶T0.2圖3 不同乳化劑配比下制備的SiO2氣凝膠微球的SEM圖片Fig.3 SEM images of the silica aerogel microspheres prepared at different emulsifier concentrations
由圖3可以看出,乳化劑配比對凝膠微球粒徑的僅有微小的影響。隨著乳化劑中Span 80的含量從80%增加到90%,SiO2氣凝膠微球的粒徑有輕微的增加;但當含量從90%增加到98%時,其大小并沒有變化。隨著乳液的親油性增大,乳液的HLB值隨之減小,就更容易形成穩定的油包水乳液。并且油包水乳液的穩定性很大程度上與乳液的HLB值有關,HLB值較大時油包水乳液的穩定性減小,則在剪切力的作用下所形成的微粒的平均粒徑相對較小。穩定的油包水乳液的HLB值通常在6以下,當復合乳化劑的HLB值過高時,乳液就變得不穩定,這對凝膠微球的形成是不利的。圖3的d, e, f分別對應的是a, b, c放大圖,由圖3可以看出凝膠微球內部是多孔結構,其由小粒子簇形成。在本實驗中,Span 80的含量為80%時溶液的HLB值為6.44,Span 80的含量為90%時溶液的HLB值為5.37,考慮到乳液穩定性,本實驗選擇Span 80 含量90%為我們大多數實驗方案。
2.2.2 攪拌速度對SiO2氣凝膠微球形貌的影響
為了研究攪拌速度對凝膠微球粒徑的影響,選擇攪拌速度從300 rpm到700 rpm。從圖4可以看出,在攪拌速度為300 rpm的條件下,凝膠微球呈現出一個較大的平均粒徑,大約為18.8 μm(見圖4(a)),隨著攪拌速度上升到400 rpm和500 rpm,SiO2氣凝膠微球的粒徑分別下降為13.6 μm(見圖4(b))和6.8 μm(見圖4(c))。當攪拌速度上升至600 rpm時,氣凝膠微球的破碎程度顯著增加(見圖4(d)),當攪拌速度達到800 rpm時,氣凝膠微球基本上完全碎裂。

(a) 300 rpm;(b) 400 rpm;(c) 500 rpm;(d) 600 rpm;(e) 700 rpm;(f) 800 rpm圖4 不同攪拌速度下制備的SiO2氣凝膠微球的SEM圖Fig.4 SEM images of the silica aerogel microspheres prepared at different stirring speed
堿性硅溶膠是由粒徑為5~100 nm的無定型SiO2在水中形成的膠體溶液,通常以氧化鈉或氨作為穩定劑,具有球形結構,其內部是由許多硅氧鍵形成的SiO2微粒,同時硅原子表面又有許多羥基[8]。所以硅溶膠具有聚合作用的特性,因此先用硝酸調節硅溶膠的pH值,然后加入一定量的乙醇將其調節成弱酸性,此時SiO2微粒在乙醇中均勻分布。將硅溶膠在攪拌的條件下滴入油相中并形成微小液滴,通過加入氨水來改變乳液的pH值使得硅溶膠在能保持其液滴的形態下發生聚合反應,以使其迅速凝膠。高速攪拌過程雖然有利于促進氨水的擴散,可使水相快速凝膠化[9~12]。另一方面,高速的攪拌過程使得輸入系統的能量變得更高,這樣就為形成新的界面提供了條件。因此,所形成的凝膠微球粒徑較小,且粒徑分布較窄。而更高的攪拌速度會使得部分凝膠的或者已經完成凝膠的微球發生變形或者破裂。
而后續的老化和干燥過程只能增強凝膠中的納米孔洞,而并不能對微米級的氣凝膠微球的形貌產生影響。因此,合適的攪拌速度對形成球狀的且粒徑分布均勻的SiO2氣凝膠微球來說是一個十分重要的影響因素。
在本實驗中,攪拌速度是一個有效的可以控制氣凝膠微球粒徑大小的因素。然而攪拌速度過高時會使得氣凝膠微球破裂。因此,在本實驗中選取攪拌速度為400 rpm。
2.3 SiO2氣凝膠微球的性能表征
2.3.1 SiO2氣凝膠微球的紅外譜圖分析
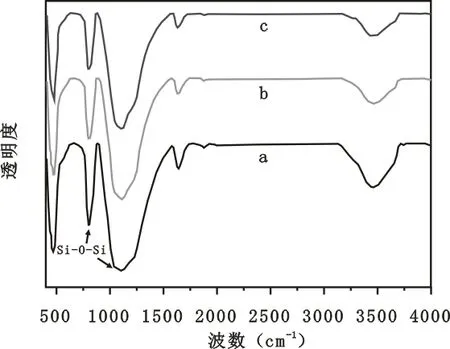
(a)SA. S8∶T2;(b)SA. S9∶T1;(c)SA. S9.8∶T0.2圖5 未改性的SiO2氣凝膠微球的紅外譜圖Fig.5 FTIR spectra of unmodified silica aerogel microspheres
圖5(a)、(b)、(c)是未改性的SiO2氣凝膠微球的紅外譜圖。發現由不同乳化劑配比所制備出的未改性的氣凝膠微球的紅外譜圖沒有差別。在3 452 cm-1的較強的吸收峰是水分子中的氫鍵的O-H伸縮振動[13]。而在1 641 cm-1的較弱的吸收峰是物理吸附水的吸收峰。在810 cm-1和1 112 cm-1的吸收峰分別對應于Si-O-Si的對稱伸縮振動和反對稱伸縮振動[14~15],這證明了SiO2網絡結構的存在。但是在其中并未發現Si-C的吸收峰,所以經過未改性的介孔SiO2氣凝膠是親水性的。
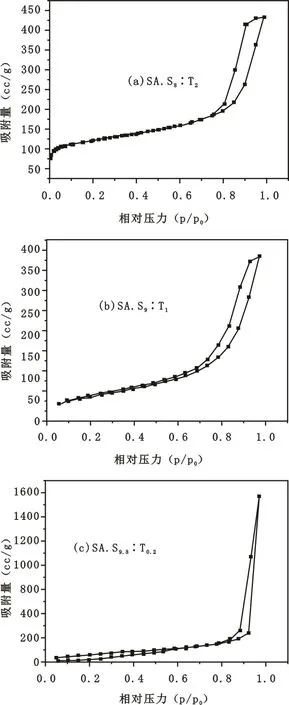
圖6 不同乳化劑配比所制備出的凝膠微球的N2吸附曲線
Fig.6 N2absorption-desorption isotherms of silica aerogel microspheres obtained with different span-80∶tween-80 mass ratio
2.3.2 SiO2氣凝膠微球的比表面積孔徑分析
圖6是不同乳化劑配比所制備出的氣凝膠微球的N2吸附曲線。
圖6中樣品S.A.S8∶T2、S.A.S9∶T1和S.A.S9.8∶T0.2的等溫線屬于典型的Type Ⅳ型等溫線,這是典型的介孔材料的等溫線[16]。吸附曲線在中間區域迅速上升并且在吸附和脫附曲線間有一個明顯的滯后環,這是由于介孔材料的毛細管凝聚現象導致的。同時從滯后環形狀的變化中可以看出孔洞形狀的變化。樣品S.A.S9.8∶T0.2的吸附-脫附支曲線在大多數p/p0的氛圍內幾乎是平行或垂直的,表明這屬于H1型滯后環,它所對應的一般是圓柱形孔,其內部具有相互連通的結構網絡[17]。樣品S.A.S8∶T2和S.A.S9∶T1吸附-脫附曲線表明其為H2型滯后環,這種類型的滯后環說明其內部結構為墨水瓶型孔洞[18]。
3 結論
筆者對乳狀液的形成及其性質進行了簡要的分析,在此基礎上對SiO2氣凝膠微球的形成機理進行了初步的探討。同時對影響SiO2氣凝膠微球形貌的因素進行了研究,得出了以下結論:
1)乳化劑配比對氣凝膠微球粒徑影響不大,但對乳液的穩定性有一定的影響;
2)攪拌速度越大,氣凝膠微球的粒徑將會逐漸減小,當攪拌速度超過600 rpm時,氣凝膠微球的破碎程度增加,當攪拌速度達到800 rpm時,已觀察不到微球的形狀;用此種方法所制備出的氣凝膠微球比表面積為207.5 m2/g±451 m2/g,密度為0.287 m3/g±0.014 g/cm3,其粒徑分布在5~20 μm,孔分布集中在20~40 nm。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
