小麥HMW-GS毛細管電泳高效分離體系研究
2018-06-13 02:12:24王衛東何慶夢姚曉露楊明明李建芳
麥類作物學報 2018年5期
王衛東,高 翔,2,何慶夢,姚曉露,劉 陽,楊明明,2,李建芳,2,楊 娜
(1.西北農林科技大學農學院,陜西楊陵 712100;2.陜西省小麥工程技術研究中心,陜西楊凌 712100;3.山西省農業科學院棉花研究所,山西運城 044000)
小麥(TriticumaestivumL.)貯藏蛋白主要包括醇溶蛋白和谷蛋白兩大類,依據分子量大小可將谷蛋白進一步分為高分子量谷蛋白亞基(HMW-GS)和低分子量谷蛋白亞基(LMW-GS)[1]。其中,HMW-GS與面筋彈性和延展性密切相關,是參與形成谷蛋白多聚體的主要成分[2];HWM-GS的組合不同則產生的品質效應不同[3]。對小麥品種中HMW-GS的分離鑒定已成為品質研究的重要環節。
傳統方法分離小麥HMW-GS多采用SDS-聚丙烯酰胺凝膠(SDS-PAGE)和高效液相色譜(HPLC)等[4-7]。SDS-PAGE分離所需時間較長,對樣品需求量較大,分離效果容易受環境及人為因素影響,且在鑒定亞基類型時需要對照“Payne命名系統”,準確性低,一些遷移率接近的亞基難以被區分。雖然結合分子標記技術可以在一定程度上彌補這一缺點,但由于特異性引物種類有限、實驗前需要對種子發苗、提取和純化樣品DNA等,工作效率較低。HPLC較 SDS-PAGE方法克服了很多不足,但其成本較高,適用范圍窄。HPCE(毛細管電泳)技術以其進樣少、運行成本低、快速、準確性高等特點,被越來越多地應用于小麥HMW-GS研究[8-11]。
HPCE中,分離介質緩沖液的選擇至關重要。目前,應用于小麥貯藏蛋白分離的常規HPCE緩沖系統主要包括磷酸鹽緩沖液系統、硼酸鹽緩沖液系統、乳酸鋁緩沖液系統及Prosot SDS。Bietz[12]最早將酸性磷酸鹽緩沖液與堿性硼酸鹽緩沖液引入到HWM-GS的HPCE中,并獲得了較好的分離效果。Werner等[13]首次使用乳酸鋁緩沖液分離小麥HMW-GS,并成功鑒定了多個品種的亞基組成,之后又以Prosort SDS為緩沖劑對HMW-GS相關特性進行了研究[14]。Sutton等[15]在Prosot SDS中加入5%甲醇,對HMW-GS的HPCE鑒定圖譜進行了研究。Bean等[16]使用Phos-gly(磷酸-甘氨酸)緩沖液對HMW-GS進行HPCE電泳分離,相比前人獲得了更好的分辨率及分離效果。
上述應用于小麥HMW-GS分離的HPCE緩沖液系統,雖然具有較好的分離效率,但重現性較差,對亞基的定性分析尚顯不足,不利于亞基標準鑒定圖譜的構建。本試驗以普通小麥中國春為標準樣,引入亞氨基二乙酸(IDA)緩沖液系統[17],擬通過研究毛細管電泳中不同緩沖液組分濃度、緩沖液pH、分離電壓、進樣時間、運行溫度、毛細管內徑等對亞基分離的影響,確立小麥HMW-GS的HPCE高效分離體系,并通過連續重復試驗、混合進樣分析對體系的分離效率及重現性進行驗證,以期為HPCE定性分析及標準鑒定圖譜的構建提供基礎。
1 材料與方法
1.1 試驗材料
小麥品種中國春、石4185、陜182,由國家小麥改良中心楊凌分中心品質實驗室提供。
1.2 方法
1.2.1 小麥HMW-GS的提取
參照文獻[7]的方法并作改進:將單粒種子研磨成粉后加1 mL 70%乙醇振蕩30 min,20 ℃ 13 000 r·min-1離心10 min,棄上清液;向沉淀中加入1 mL提取液A(50%異丙醇),混勻,60 ℃水浴30 min;室溫下13 000 r·min-1離心10 min,棄上清,重復此步驟2次。向沉淀中加入150 μL提取液B[50%異丙醇+3 mol·L-1Tris-HCl(pH=8.8)+25 g·L-1二硫蘇糖醇],混勻后60 ℃水浴30 min。加入150 μL提取液C[50%異丙醇+3 mol·L-1Tris-HCl(pH=8.8)+10 g·L-14-乙烯基吡啶],65 ℃繼續水浴1 h。將水浴后產物于20 ℃ 13 000 r·min-1離心15 min,取120 μL上清液,加入80 μL預冷(-20 ℃)的丙酮溶液,使丙酮占總體積的40%。將混合液于-20 ℃冰箱中沉淀過夜,20 ℃ 13 000 r·min-1離心10 min,棄上清液,所得沉淀即為HMW-GS。
1.2.2 毛細管電泳系統運行前后處理
將1.2.1所得的蛋白沉淀烘干,加入200 μL蛋白溶解液(每50 mL含:15 mL丙三醇+18 g尿素+75 μL乙酸),于35 ℃水浴溶解1 h,-20 ℃保存備用,使用前超聲脫氣。與毛細管電泳相關的緩沖液及沖洗試劑均由超純水配置,經0.45 μm濾膜過濾,20 ℃、50 W超聲脫氣15 min。
電泳分離前的沖洗程序為:N2反向吹干1 min,1 mol·L-1H3PO4沖洗2 min,0.1 mol·L-1NaOH沖洗2 min,去離子水沖洗1 min,緩沖液沖洗3 min。實驗結束后用0.1 mol·L-1NaOH沖洗2 min,反向去離子水沖洗1 min。
1.2.3 毛細管電泳分離體系的優化
使用P/ACE MDQ plus毛細管電泳儀(Beckman,USA)對樣品進行分析。毛細管配置為檢測長度27 cm的石英非涂層毛細管(USA)。IDA緩沖液組分為:IDA(亞氨基二乙酸)+HPMC(羥丙基甲基纖維素)+ACN(乙腈)。利用控制變量法,對緩沖液系統及電泳分離參數進行逐一優化,確立HMW-GS毛細管分離最佳體系。緩沖液系統pH設置為2.2、2.5、2.8;緩沖液組分中IDA濃度設置為50、75、100 mmol·L-1;HPMC濃度設置為0、0.05%、0.5%;ACN濃度設置為10%、15%、20%。電泳參數中,分離電壓設置為15、20、25 kV;運行溫度設置為25、30、35 ℃;毛細管內徑設置為25、50 μm;進樣時間設置為5、8 s。
1.2.4 HMW-GS毛細管電泳分離體系重現性驗證
利用構建好的HPCE高效分離體系,對小麥品種中國春的HMW-GS進行連續30次及連續多天電泳分離,檢驗毛細管電泳圖峰形、基線穩定性。將中國春與石4185、陜182混合進樣,比較該體系與常規Phos-gly/ACN體系(磷酸-甘氨酸+HPMC+ACN)的分離效率及圖譜分辨率,計算各亞基出峰時間的RSD值,驗證分離重現性。
2 結果與分析
2.1 緩沖液系統pH的選擇
依據Salmanowicz等[18]、Yan等[19]的研究,毛細管電泳分離圖譜中,中國春的HMW-GS具有四個特征峰,按遷移時間順序分別為1Dy12→1By8→1Bx7→1Dx2。由圖1可知,在緩沖液為100 mmol·L-1IDA+0.05% HPMC+20% ACN,采用25 μm內徑毛細管,214 nm檢測波長,分離電壓15 kV,運行溫度25 ℃,10.0 kV電進樣5 s條件下,緩沖液pH對HMW-GS峰高、峰寬及基線均有顯著影響。pH為2.8時,8.13 min出現1Dy12亞基主峰;1By8主峰出峰時間為9.97 min,峰高較低,主峰辨別度差,且峰后有基線的波動;1Bx7主峰出峰時間為11.54 min;1Dx2主峰出現在13.12 min;從1Dy12到1Dx2亞基峰時間跨度為4.99 min;整體上看基線有明顯的上升趨勢。pH為2.5時,四個主峰出峰時間分別為10.24、11.94、13.40和15.00 min;相比pH2.8時出峰時間較晚,時間跨度稍有減小(4.76 min),但整體峰形對稱、分離度較高、基線相對平穩。pH為2.2時,四個主峰出峰時間為11.57、12.55、14.31和16.08 min,時間跨度為4.51 min;相比pH為2.5、2.8,峰寬度增大、分離度降低;而且在臨近1Dy12亞基主峰時基線波動較大,1By8主峰與1Bx7主峰之間也出現了類似現象。說明pH為2.5時亞基分離效果較好。

A、B、C曲線所對應的pH分別為2.8、2.5、2.2。
The corresponding pH of A,B and C curves are 2.8,2.5 and 2.2,respectively.
圖1不同pH緩沖液對HMW-GS電泳分離的影響
Fig.1EffectofdifferentpHofbufferonelectrophoresisseparationofHMW-GS
2.2 緩沖液系統組分濃度的選擇
2.2.1 IDA濃度的選擇
如圖2所示,緩沖液為:0.05% HPMC+20% ACN,pH 2.5。電泳條件為:毛細管內徑25 μm,檢測波長214 nm,分離電壓15 kV,運行溫度25 ℃,10.0 kV進樣5 s,中國春的HMW-GS亞基整體遷移速率隨IDA濃度的增大而加快,濃度對亞基峰高、峰形及基線狀況影響不大。比較相同IDA濃度下連續兩針的電泳圖譜發現,IDA濃度對電泳分離重現性的影響較大。IDA濃度為50 mmol·L-1時,亞基遷移速率最慢,電泳第一針與第二針之間的出峰時間差異最大。IDA濃度為100 mmol·L-1時,亞基遷移最快,連續兩針間電泳出峰時間差值減小,但仍然較大,且在1Dy12主峰之前出現了基線的波動。IDA濃度為75 mmol·L-1時,亞基遷移速率介于50和100 mmol·L-1之間,兩針間電泳出峰時間幾乎重合,亞基峰形、基線均保持較高的一致性,電泳分離重現性較高。說明IDA濃度為75 mmol·L-1的亞基分離效果較好。

A和B、C和D、E和F曲線分別表示IDA濃度為100、75、50 mmol·L-1時電泳第一針、第二針。
A and B,C and D,and E and F represent the first and second electrophoresis when the concentration of IDA was 100 mmol·L-1,75 mmol·L-1and 50 mmol·L-1,respectively.
圖2不同IDA濃度對HMW-GS電泳分離的影響
Fig.2EffectofdifferentconcentrationofIDAontheelectrophoresisseparationofHMW-GS
2.2.2 HPMC濃度的選擇
如圖3所示,緩沖液為75 mmol·L-1IDA+20% ACN,pH 2.5。電泳條件為:毛細管內徑25 μm,檢測波長214 nm,分離電壓15 kV,運行溫度25 ℃,10.0 kV進樣5 s。HPMC濃度對HMW-GS分離度及基線狀況有顯著影響。不添加HPMC時,亞基遷移速度最快,但峰之間未完全分離,主峰界線不明顯,且峰形不規則。HPMC濃度為0.5%時,峰寬明顯變窄,亞基分離度最高,但基線噪音較大,影響了圖譜的分辨率。HPMC濃度為0.05%時,1Dy12、1By8、1Bx7、1Dx2四個主峰出峰時間分別為12.40、14.07、15.54和17.16 min,亞基分離度介于0與0.5%濃度之間,雖然亞基遷移速率降低,但主峰峰形對稱,且基線噪音很小,圖譜分辨率高。說明HPMC濃度為0.05%時最佳。
2.2.3 ACN濃度的選擇
緩沖液為75 mmol·L-1IDA+0.05 % HPMC,pH 2.5,毛細管內徑25 μm,檢測波長214 nm,分離電壓15 kV,運行溫度25 ℃,10.0 kV進樣5 s。添加不同濃度ACN,對HMW-GS毛細管電泳分離的影響主要集中在主峰分離度上(圖4)。ACN濃度為20%時,四個主峰分離度最低。ACN濃度為10%時,主峰分離度最大,但是峰寬增大、峰高明顯降低,即亞基縱向擴散與峰展寬比例減小,影響圖譜的分辨率。當ACN濃度為15%時,主峰分離度介于前兩者之間,1Dy12、1By8、1Bx7、1Dx2主峰依次出現在12.68、14.32、16.80和18.94 min,亞基峰形清晰對稱,基線噪音最低。因此,ACN濃度為15%最佳。

A、B、C曲線分別對應HPMC濃度0.5%、0.05%、0。
A,B and C curves correspond to 0.5%,0.05% and 0 of HPMC,respectively.
圖3不同HPMC濃度對HMW-GS電泳分離的影響
Fig.3EffectofdifferentconcentrationofHPMConthe
electrophoresisseparationofHMW-GS

A:20% ACN; B:15% CAN; C:10% ACN.圖4 不同ACN濃度對HMW-GS電泳分離的影響Fig.4 Effect of different concentration of ACN on theelectrophoresis separation of HMW-GS
2.3 毛細管電泳分離條件的選擇
2.3.1 毛細管分離電壓的選擇
利用已經優化好的IDA緩沖液系統,對毛細管電泳運行參數進行逐步優化,進一步提高HMW-GS分離效果。
緩沖液為75 mmol·L-1IDA+0.05% HPMC+15% ACN,pH為2.5;毛細管內徑25 μm,檢測波長214 nm,運行溫度25 ℃,10.0 kV進樣5 s。如圖5所示,當分離電壓為15 kV時,基線水平、亞基分離度較高,但是峰高相對較低,且遷移速度最慢。當電壓增大為20 kV時,主峰分離度未受明顯影響,與15 kV差異不大,亞基峰高明顯增大、整體遷移速率明顯加快,圖譜分辨率得到了提升。電壓達到25 kV時,雖然獲得了最大遷移速率及峰高,但是基線出現了整體下降的趨勢。由此可見,分離電壓的提高對遷移速率及峰高有促進作用,但會增加分離不穩定因素。綜合來看,20 kV分離電壓對HMW-GS的分離效果最佳。

A:15 kV; B:20 kV; C:25 kV.圖5 分離電壓對HMW-GS電泳分離的影響Fig.5 Effect of separation voltage on the electrophoresisseparation of HMW-GS
2.3.2 分離溫度的選擇
緩沖液為75 mmol·L-1IDA+0.05% HPMC+15% ACN,pH 2.5;毛細管內徑25 μm,檢測波長214 nm,分離電壓20 kV,10.0 kV進樣5 s。當分離溫度為25 ℃時,圖形清晰,亞基峰形對稱,四個主峰出峰時間依次為10.93、12.81、15.98和18.13 min(圖6)。溫度為30 ℃時,亞基分離度與25 ℃時接近,但亞基出峰更早;四大主峰分別出現在9.31、11.69、13.18和14.77 min,時間跨度為52.4 min,整體遷移速度更快;亞基縱向擴散與峰展寬比例擴大,從而帶來主峰辨識度的提高。溫度為35 ℃時亞基分離度達到最大,出峰時間分別為8.22、10.38、13.14和15.56 min,但是亞基峰高明顯變小,基線波動較大,噪音增強,導致圖譜分辨率明顯下降。由此可見,分離溫度為30 ℃時分離效果最好。
2.3.3 PDA檢測波長的選擇
緩沖液為75 mmol·L-1IDA+0.05% HPMC+15% ACN,pH 2.5;毛細管內徑25 μm,分離電壓20 kV,運行溫度30 ℃,10.0 kV進樣5 s。結果(圖7)表明,PDA檢測波長與分離圖譜中亞基峰寬和基線噪音密切相關,對峰高有輕微影響,對圖譜亞基出峰時間的影響不大。PDA波長由214 nm轉變為200 nm時,基線噪音更小,亞基峰寬減小,峰高略微增大,這使得亞基縱向擴散與峰展寬比例進一步增大,帶來了主峰辨識度的提高。因此PDA檢測波長為200 nm時最佳。

A、B、C曲線分別對應25、30和35 ℃。
A,B and C curves are 25,30 and 35 ℃,respectively.
圖6毛細管內運行溫度對HMW-GS電泳分離的影響
Fig.6Effectofthecapillarytemperatureonthe
electrophoresisseparationofHMW-GS

A:214 nm; B:200 nm.圖7 PDA檢測波長對HMW-GS電泳分離圖譜的影響Fig.7 Effect of PDA wavelengths on theelectrophoresis separation of HMW-GS
2.3.4 進樣時間的選擇
緩沖液為75 mmol·L-1IDA+0.05% HPMC+15% ACN,pH 2.5;毛細管內徑25 μm,檢測波長200 nm,分離電壓20 kV,運行溫度30℃,10.0 kV進樣。當進樣時間為5 s時,峰形清晰,亞基分離度高,基線平穩。進樣時間為8 s時,電泳遷移速率與進樣5 s差異不大,但峰寬明顯增大,1Bx7、1Dx2主峰峰高顯著降低,四個主峰之間界線模糊,亞基分離度較差,圖譜分辨率降低(圖8)。因此,進樣時間為5 s時分離效果好。

A:8 s; B:5 s.圖8 不同進樣時間對HMW-GS電泳分離圖譜的影響Fig.8 Effect of different sampling time on theelectrophoresis separation of HMW-GS
2.3.5 毛細管內徑的選擇
緩沖液為75 mmol·L-1IDA+0.05% HPMC+15% ACN,pH 2.5;檢測波長200 nm,分離電壓20 kV,運行溫度30 ℃,10.0 kV進樣5 s。結果(圖9)表明,毛細管內徑同時影響亞基峰寬、分離度及基線穩定性。毛細管內徑為25 μm時,亞基分離度高,亞基峰形對稱,基線水平,背景噪音低。當毛細管內徑為50 μm時,亞基分離度降低,峰寬增大,峰高降低;電泳遷移率降低,亞基出峰較晚,1Dy12、1By8、1Bx7、1Dx2四個主峰依次出現在第10.81、12.24、13.78和15.58 min處;1Dy12主峰出峰之前有明顯的基線波動,電泳分離后期基線逐漸上升,且有倒峰及雜峰出現。由此可見,毛細管內徑為25 μm時最佳。
2.4 HMW-GS毛細管電泳高效分離體系及驗證
在優化后條件下,即緩沖液組分為15% ACN+75 mmol·L-1IDA+0.05% HPMC,pH為2.5;毛細管內徑25 μm, PDA檢測波長200 nm,分離電壓20 kV,運行溫度30 ℃,樣品10.0 kV電進樣5 s,對中國春的HMW-GS連續多次電泳分離。結果(圖10)發現,第1~2 min為系統峰(溶劑峰),3~6 min為微量LMW-GS區域,HMW-GS在9 min后開始出峰,1Dy12、1By8、1Bx7、1Dx2亞基16 min內全部分離完成。連續30次、連續6 d重復試驗(圖10、11),均顯示圖譜亞基峰形一致、分離度高、基線水平。通過混合進樣方式,與常規的Phos-gly/ACN體系進行對比。

A:25 μm; B:50 μm.圖9 不同毛細管內徑對HMW-GS電泳分離圖譜的影響Fig.9 Effect of different inner diameters of the capillaryon the electrophoresis separation of HMW-GS

圖中所示曲線從下往上依次為第1、5、10、15、20、25、30次電泳分離曲線。
The curves from bottom to top are first,fifth,tenth,fifteenth,twentieth,twenty-fifth and thirtieth electrophoresis separation curves,respectively.
圖10中國春HMW-GS連續30次毛細管電泳分離圖譜
Fig.10Imageofthirtyconsecutiveinjections
ofHMW-GSofChineseSpring
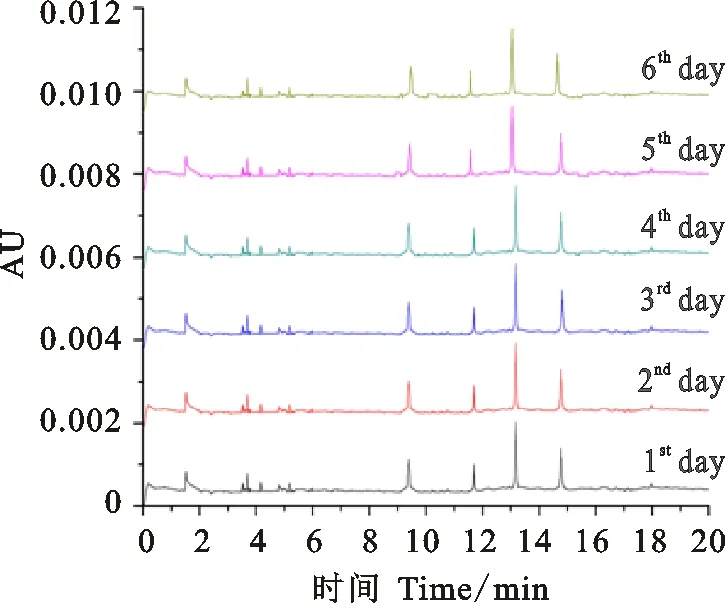
圖中所示曲線從下往上依次為第1、2、3、4、5、6天電泳分離曲線。
The curves from bottom to top are the curves of the first day,the second day,the third day,the fourth day,the fifth day and the sixth day,respectively.
圖11中國春HMW-GS連續6d電泳圖譜
Fig.11ContinuousrepeatedexperimentofHMW-GSofChineseSpringforsixdays
結果(圖12、表1)表明,該體系在亞基分離速度、圖譜分辨率上更具有優勢,對于遷移率比較接近的亞基(如1Dy12與1Dy10、1By9與1By8、1Dx5與1Dx2)更容易表征;亞基出峰時間RSD(<0.2%)值遠小于后者,說明分離重現性更高。

A、B均為中國春(Null,7+8,2+12)與石4185(1,7+9,2+12)、陜182(1,7+8,5+10)混合樣的分離圖譜。圖A電泳條件:25 μm毛細管,200 nm檢測波長,緩沖液組分75 mmol·L-1IDA+0.05% HPMC+15% ACN,pH 2.5,分離電壓20 kV,運行溫度30 ℃,10.0 kV電進樣5 s。圖B電泳條件為:25 μm毛細管,214 nm檢測波長,緩沖液組分100 mmol·L-1Phosphate-glycine+0.05% HPMC+20% ACN,pH 2.5,分離電壓12.5 kV,運行溫度40 ℃,10.0 kV電進樣5 s。
A and B are both the separation curves obtained from the mixture of Chinese Spring(Null,7+8,2+12) ,Shi 4185(1,7+9,2+12) and Shaan 182(1,7+8,5+10).The electrophoresis conditions of Fig.A:Inner diameter of the capillary was 25 μm; Detection wavelength was 200 nm; Buffer composition was 75 mmol·L-1IDA + 0.05% HPMC + 15% ACN,with pH at 2.5; Separation voltage was 20 kV; Operating temperature was 30 ℃; Samples were injected for five seconds by electric method under under 10.0 kV.The electrophoresis conditions of Fig.B:Inner diameter of the capillary was 25 μm;Detection wavelength was 214 nm; Buffer composition was 100 mmol·L-1phosphate-glycine + 0.05% HPMC + 20% ACN,with pH at 2.5;Separation voltage was 12.5 kV;Operating temperature was 40 ℃;Samples were injected for five seconds by electric method under under 10.0 kV.

圖12 不同分離體系效果比較
表中數據為連續30次電泳分離所得平均值。
The data is the average value obtained from 30 consecutive injections.
3 討 論
小麥HMW-GS與小麥品質密切相關,其分離鑒定是品質研究的重要環節。與傳統方法相比,HPCE具有快速、自動化、分辨率高等特點。依據電泳圖譜可獲得亞基的準確峰高、遷移時間等,有助于亞基定性及定量分析。本試驗結果表明,HPCE分離體系中緩沖液pH、緩沖液組分、分離電壓、運行溫度、進樣時間、檢測波長及毛細管內徑等條件的變化均會影響分離效果。
Bean等[20]最先將IDA/ACN緩沖系統應用到蛋白HPCE分析中,主要對醇溶蛋白的分離進行了探討,本試驗首次將其引入到HMW-GS的HPCE分析中,通過優化條件,同樣獲得了良好的分離效果。Righetti等[21]表明IDA/ACN緩沖液系統分離蛋白質的最佳pH為2.2~2.8。本研究發現,pH > 2.5會導致基線不穩定趨勢增加。由于HMW-GS屬于大分子蛋白[22],容易吸附在毛細管壁上,使電滲流改變,影響電泳分離重復性,極端pH可以抑制蛋白吸附,維持電滲流穩定,但當pH達到一定范圍時,該作用將顯著減弱。推測pH>2.5時抑制作用降低,電泳后期HMW-GS吸附作用增強,從而產生了較多的焦耳熱,導致了基線的上升。IDA在毛細管電泳中主要影響分離的重現性,本研究結果表明,其濃度為75 mmol·L-1時,分離體系重現性較好。HPMC是一種親水高分子聚合物[23],在緩沖液系統中具有充當篩分介質[24]和動態修飾的作用[25],影響亞基分離度及基線狀況。HPMC濃度增大可能提高毛細管電泳靈敏度,但相應的圖譜噪音也會增強。適當降低濃度可增大亞基分離度。由于緩沖液中HPMC濃度的改變亦可影響其粘性,緩沖介質粘性是影響電滲流的主要因素之一[26],因此控制HPMC濃度有助于穩定毛細管電泳電滲流。ACN在緩沖液系統中的作用是通過加快樣品堆積,促進遷移速率[27],其濃度的變化往往影響電泳靈敏度,本試驗發現,當ACN濃度提高時圖譜噪音也會相應增高,再次驗證了這一觀點。由于HPCE是以高壓電場為驅動力,溫度影響著管內溶液的粘性[28],因此兩者數值升高均會提高圖譜遷移速率,但也為電泳系統帶來不穩定因素。本試驗中,PDA檢測波長對分離效果的影響主要是在圖譜分辨率上,由于不同物質的最大吸收波長不同[29],改變PDA檢測波長則會引起圖譜信噪比的變化。電壓進樣時間往往會影響進樣塞的長度,進樣塞長度過大時容易使峰展寬大于縱向擴散[30],與本試驗結果一致。本試驗還發現,增大毛細管內徑,會使分離度降低、基線波動,這可能是因為毛細管作為分離通道,其內徑對散熱影響較大;減小其內徑有利于電泳過程中熱量的散出,減少焦耳熱的產生;內徑過大,容易在毛細管內部形成溫度梯度(中心溫度高),破壞塞流,進而導致亞基縱向擴散小于區帶展寬[31],降低分離效率。
本研究通過分析HPCE中不同因素對HMW-GS分離效果的影響,結合連續重復實驗及比對驗證,成功構建了基于IDA緩沖液系統的HPCE高效分離體系,由于該體系下圖譜亞基分離度高、重現性好,因此有利于HMW-GS的定性分析。結合SDS-PAGE及分子標記等,可對HPCE分離圖譜中不同類型HMW-GS進行表征,確定標準遷移時間。依據標準遷移時間,即可完成小麥材料中相關HMW-GS的快速鑒別。同時,該指標亦可作為新型未知亞基的鑒定標準。
參考文獻:
[1] SHEWRY P R,TATHAM A S,LAZZERI P.Biotechnology of wheat quality [J].JournaloftheScienceofFoodandAgriculture,1997,73(4):399.
[2] PAYNE P I,NIGHTINGALE M A,KRATTIGER A F,etal.The relationship between HMW glutenin subunit composition and the bread-making quality of British-grown wheat varieties [J].JournaloftheScienceofFoodandAgriculture,1987,40(1):64.
[3] WIESER H,ZIMMERMANN G.Importance of amounts and proportions of high molecular weight subunits of glutenin for wheat quality [J].EuropeanFoodResearch&Technology,2000,210(5):329.
[4] VISIOLI G,COMASTRI A,IMPERIALE D,etal.Gel-based and gel-free analytical methods for the detection of HMW-GS and LMW-GS in wheat flour [J].Foodanalyticalmethods,2016,9(2):474.
[5] HAJAS L,SCHERF K A,TRK K,etal.Variation in protein composition among wheat(TriticumaestivumL.) cultivars to identify cultivars suitable as reference material for wheat gluten analysis [J].FoodChemistry,2017,5:5.
[6] HOSSEIN M,REZA M.Characterization of wheat gluten subunits by liquid chromatography ? Mass spectrometry and their relationship to technological quality of wheat [J].JournalofCerealScience,2017,76:233.
[7] JANG Y R,BEOM H R,ALTENBACH S B,etal.Improved method for reliable HMW-GS identification by RP-HPLC and SDS-PAGE in common wheat cultivars [J].Molecules,2017,22(7):1055.
[8] JIANG P,GAO J,ZHENG X,etal.Clustered transcription initiators and expression of HMW-GS genes in wheat endosperm [J].CropScience,2017,57(1):384.
[9] LOOKHART G L,BEAN S R,JONES B L.Separation and characterization of barley(HordeumvulgareL.) hordeins by free zone capillary electrophoresis [J].Electrophoresis,1999,20(7):1611.
[10] LOOKHART G L,BEAN S R.Improvements in cereal protein separations by capillary electrophoresis:Resolution and reproducibility [J].CerealChemistry,1996,73(1):85.
[11] LOOKHART G,BEAN S.Separation and characterization of wheat protein fractions by high-performance capillary electrophoresis [J].CerealChemistry,1996,72(6):530.
[12] BIETZ J A.Fractionation of wheat gluten proteins by capillary electrophoresis [J].GlutenProteins,1993:406.
[13] WERNER W E,WIKTOROWICZ J E,KASARDA D D.Wheat varietal identification by capillary electrophoresis of gliadins and high molecular weight glutenin subunits [J].CerealChemistry,1994,71(5):400.
[14] WERNER W E.Ferguson plot analysis of high molecular weight glutenin subunits by capillary electrophoresis [J].CerealChemistry,1995,72:248.
[15] SUTTON K H,BIETZ J A.Variation among high molecular weight subunits of glutenin detected by capillary electrophoresis [J].JournalofCerealScience,1997,25(1):9.
[16] BEAN S R,LOOKHART G L.Faster capillary electrophoresis separation of wheat proteins through modifications to buffer composition and sample handling [J].Electrophoresis,1998,19(18):3191.
[17] LOOKHART G L,BEAN S R.Ultrafast CE analysis of cereal storage proteins and its applications to protein characterization and cultivar differentiation [J].JournalofAgriculturalandFoodChemistry,2000,48:353.
[18] SALMANOWICZ B P,LANGNER M,FRANASZEK S.Charge-based characterisation of high-molecular-weight glutenin subunits from common wheat by capillary isoelectric focusing [J].Talanta,2014,129:14.
[19] YAN Y,JIANG Y,SUN M,etal.Rapid identification of HMW glutenin subunits from different hexaploid wheat species by acidic capillary electrophoresis [J].CerealChemistry,2004,81(5):565.
[20] BEAN S R,LOOKHART G L.Ultrafast capillary electrophoretic analysis of cereal storage proteins and its applications to protein characterization and cultivar differentiation [J].JournalofAgriculturalandFoodChemistry,2000,48(2):350.
[21] RIGHETTI P G,OLIVIERI E,VIOTTI A.Identification of maize lines via capillary electrophoresis of zeins in isoelectric,acidic buffers [J].Electrophoresis,1998,19(10):1740.
[22] DENG Z,TIAN J,LIU X.Accumulation regularity of protein components in wheat cultivars with different HMW-GS [J].ActaAgronomicaSinica,2004,30(5):484.
[23] SARKAR N,WALKER L C.Hydration-dehydration properties of methylcellulose and hydroxypropyl methylcellulose [J].CarbohydratePolymers,1995,27(3):183.
[24] ORIARD M.Separation of proteins with capillary electrophoresis in coated capillaries with and without electroosmosis studies on zone broadening and analytical performances [J].ActaUniversitatisUpsaliensis,2006,19(1):50.
[25] MACHISTE E O,BUCKTON G.Dynamic surface tension studies of hydroxypropylmethylcellulose film-coating solutions [J].InternationalJournalofPharmaceutics,1996,145(1-2):197.
[26] YIN S,SU M,XIE G,etal.Factors affecting separation and detection of bile acids by liquid chromatography coupled with mass spectrometry in negative mode [J].AnalyticalandBioanalyticalChemistry,2017:6.
[27] STEFAN A R,DOCKERY C R,NIEUWLAND A A,etal.Forensic analysis of anthraquinone,azo,and metal complex acid dyes from nylon fibers by micro-extraction and capillary electrophoresis [J].Analytical&BioanalyticalChemistry,2009,394(8):2081.
[28] 陳 義.毛細管電泳技術及應用[M].北京:化學工業出版社,2000:15.
CHEN Y.Technology and application of capillary electrophoresis [M].Beijing:Chemical Industry Press,2000:15.
[29] POTIER I L,BOUTONNET A,ECOCHARD V,etal.Chemical and instrumental approaches for capillary electrophoresis(CE)-fluorescence analysis of proteins [M]//Nguyet Thay Tran,Myrian Tavema.Capillary Electrophoresis of Proteins and Peptides.Nwe York:Humana Press,2016:1-10.
[30] WATZIG H,DETTE C.Capillary electrophoresis(CE)-a review.Strategies for method development and applications related to pharmaceutical and biological sciences [J].DiePharmazie,1994,49(2-3):93.
[31] WESTON A,BROWN P R,JANDIK P,etal.Factors affecting the separation of inorganic metal cations by capillary electrophoresis [J].JournalofChromatographyA,1992,593(1):294.
