實時熒光定量PCR技術在實驗教學中的應用
2018-04-25 07:43:22趙玉紅趙立青李小菊石建黨李登文張金紅
實驗技術與管理 2018年4期
趙玉紅, 李 欣, 趙立青, 李小菊, 石建黨, 李登文, 張金紅, 周 浩
(南開大學 生物實驗教學中心, 天津 300071)
實時熒光定量PCR技術(real-time fluorescent quantitative PCR,RT-qPCR)是在傳統PCR(聚合酶鏈式反應)技術基礎上發展起來的一種高度靈敏的核酸定量技術[1],具有快速、靈敏、特異性強、污染少且能實時監測等優點,被廣泛應用于mRNA表達分析、基因定量分析、點突變分析、等位基因分析、單核苷酸多態性分析、DNA甲基化的檢測及對多種傳染病病原體的定量定性分析等[2]。其基本工作原理是:在PCR反應體系中加入熒光基團,利用熒光信號積累來實時監測整個PCR進程,得到產物擴增曲線,然后采用合適的數據分析方法,計算初始模板數量(絕對定量)或初始模板數量的相對比值(相對定量)[3]。
為使學生掌握這項在生命科學中廣泛應用的先進核酸定量分析技術,設計“實時熒光定量PCR分析IPTG誘導EGFP基因在大腸桿菌中的異源表達”綜合實驗。將EGFP基因插入原核表達載體pET-28a中,轉化至大腸桿菌BL21。以IPTG(異丙基硫代β-D-半乳糖苷)不同的誘導濃度、不同的誘導溫度對增強型綠色熒光蛋白(EGFP)基因進行誘導表達,分析IPTG不同誘導濃度、不同誘導溫度對蛋白表達量的影響,以確定最佳表達條件。
1 實驗材料與儀器
菌株:含pET-28a-EGFP質粒的大腸桿菌BL21。
主要試劑:細菌RNA提取試劑盒(Omega),RevertAid First Strand cDNA Synthesis Kit(Thermo),熒光定量預混試劑盒(SuperReal,TIANGEN),LB培養基以及巰基乙醇、IPTG等。
主要儀器:實時熒光定量PCR儀(Bio-Rad-IQ5),高速冷凍離心機(Beckman J-25),金屬浴(卡尤迪),渦旋混合儀(上海滬西-WH-2)以及超微量分光光度計(Nanodrop2000c)等。
2 實驗方法
2.1 大腸桿菌誘導培養及菌體收集
(1) 大腸桿菌誘導培養:在LB培養基中加入Amp(氨芐青霉素)至終質量濃度為100 mg/L,37 ℃、200 r/min振蕩培養過夜;將過夜培養的大腸桿菌按1%的接種量接種于新的LB培養基(Amp終質量濃度為100 mg/L),37 ℃、200 r/min培養,OD600至0.6~0.8。
(2) 菌體收集:分裝菌液,共10份,每份10 mL,按照表1添加一系列濃度的IPTG,分別于16 ℃誘導培養18 h、37 ℃誘導培養3 h。每3 mL收集一管菌體,可直接提取總RNA或-80 ℃保存備用。
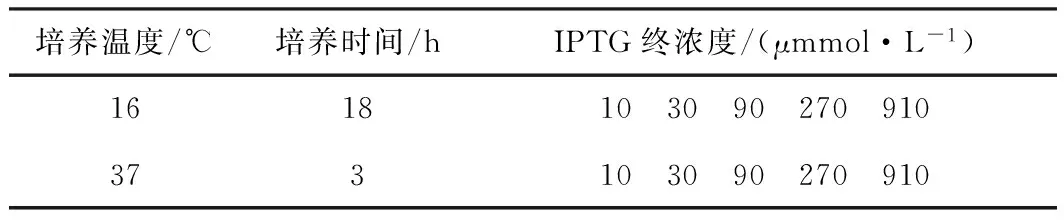
表1 IPTG不同濃度、不同溫度誘導培養下的大腸桿菌
2.2 大腸桿菌總RNA提取
參照試劑盒說明書。取3 mL菌液,4 ℃,5 000×g、離心6 min,去上清;加入100 μL溶菌酶/TE buffer,渦旋30 s,30 ℃、溫育10 min;加入350 μL BRK和30 mL beads,充分渦旋5 min,13 000g、離心5 min;取400 μL上清轉移至1.5 mL新管,70 ℃溫育5 min,13 000g、離心2 min;取上清轉移至新管,加入280 μL無水乙醇,劇烈渦旋15 s;將所有樣品轉移至2 mL離心管的吸附柱內,9 000g、離心30 s;加入400 μL RNA洗脫bufferⅠ,10 000g、離心2 min,棄洗脫液和收集管;加入500 μL洗脫bufferⅡ,9 000g、離心30 s,棄洗脫液;9 000g、空離2 min;轉移柱子至1.5 mL新管,加60 μL DEPC水,9 000g、離心1 min,重復1次。以DEPC水作為參照,用超微量分光光度計檢測RNA濃度及其純度。
2.3 反轉錄-PCR
以Oligo(dT)18作為引物,參照RevertAid First Strand cDNA Synthesis Kit試劑盒使用說明書,按表2配制反轉錄體系,充分混勻,瞬時離心。每個樣品做3個平行,陰性對照不加模板RNA。RT-PCR反應條件:25 ℃,5 min;42 ℃,60 min;70 ℃,5 min;4 ℃,Forever。所得cDNA可直接用于實時熒光定量PCR或-80 ℃保存備用。

表2 逆轉錄合成cDNA的體系
2.4 實時熒光定量PCR
2.4.1 PCR特異性引物的設計和合成
利用NCBI 上Gen Bank數據庫,查詢EGFP和大腸桿菌16S rRNA基因的mRNA序列,利用Primer 5.0軟件設計引物,并用BLAST進行同源性比較,按引物設計原則和 BLAST 同源性比較結果,確定目的片段序列及長度。引物由華大基因合成。
16SrRNA-up1:CTTGCTGCTTTGCTGACGAG
16SrRNA-dn1:GGTCCCCCTCTTTGGTCTTG
擴增片段長度為125 bp。
EGFP-up1:CAAGATCCGCCACAACATCG
EGFP-dn1:GACTGGGTGCTCAGGTAGTG
擴增片段長度為120 bp。
2.4.2 反應體系
反應體系見表3。
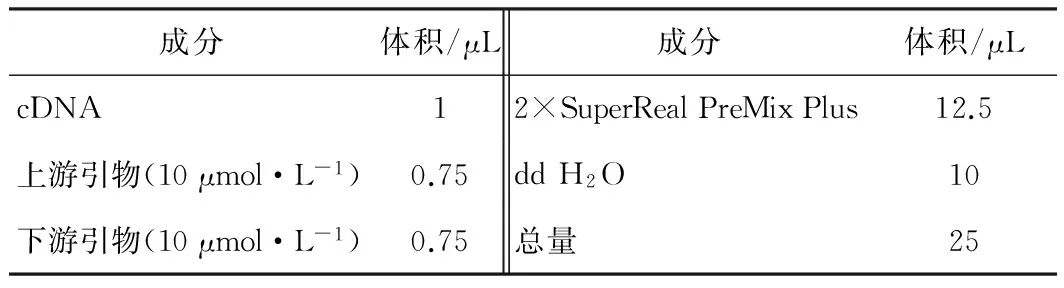
表3 實時熒光定量PCR反應體系
2.4.3 反應程序
熒光定量擴增反應程序:(1)95 ℃預變性5 min;(2)95 ℃變性10 s;(3)60 ℃復性10 s;(4)72 ℃延伸20 s,(2)—(4)進行40個循環;(5)在反應程序結束后,以0.5 ℃/5 s的速率從60 ℃緩慢遞增到95 ℃,連續測定樣品的熒光強度,進行熔解曲線分析。
2.4.4 統計學分析
用Excel 建立數據庫,應用SPSS17.0 統計軟件包進行統計分析,組間比較采用方差分析檢驗,以p<0.05 為差異有統計學意義。
3 結果與分析
3.1 大腸桿菌總RNA提取
大腸桿菌中總RNA的提取結果見表4。結果顯示,用試劑盒法提取的RNA濃度較高,同時A260/A280比值在1.8~2.2之間,表明大腸桿菌總RNA提取質量高,可用于后續的實時熒光定量PCR分析。

表4 大腸桿菌總RNA提取結果
3.2 擴增曲線
熒光定量PCR擴增結果見圖1,擴增曲線呈現出熒光背景信號階段、熒光信號指數擴增階段和平臺期3個階段,S型曲線光滑平穩,符合定量檢測要求[4]。
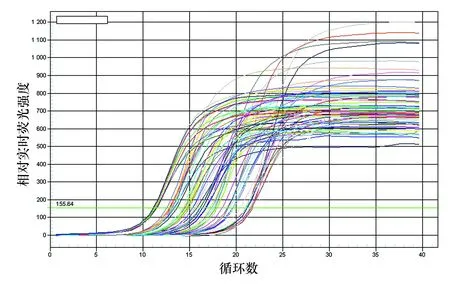
圖1 擴增曲線
3.3 熔解曲線
熔解曲線結果見圖2,擴增產物為單一峰,對應目的基因EGFP的Tm(DNA溶解溫度)值均一,未出現非特異性擴增和明顯引物二聚體。因此,可以采用SYBR GreenⅠ法進行相對定量分析。
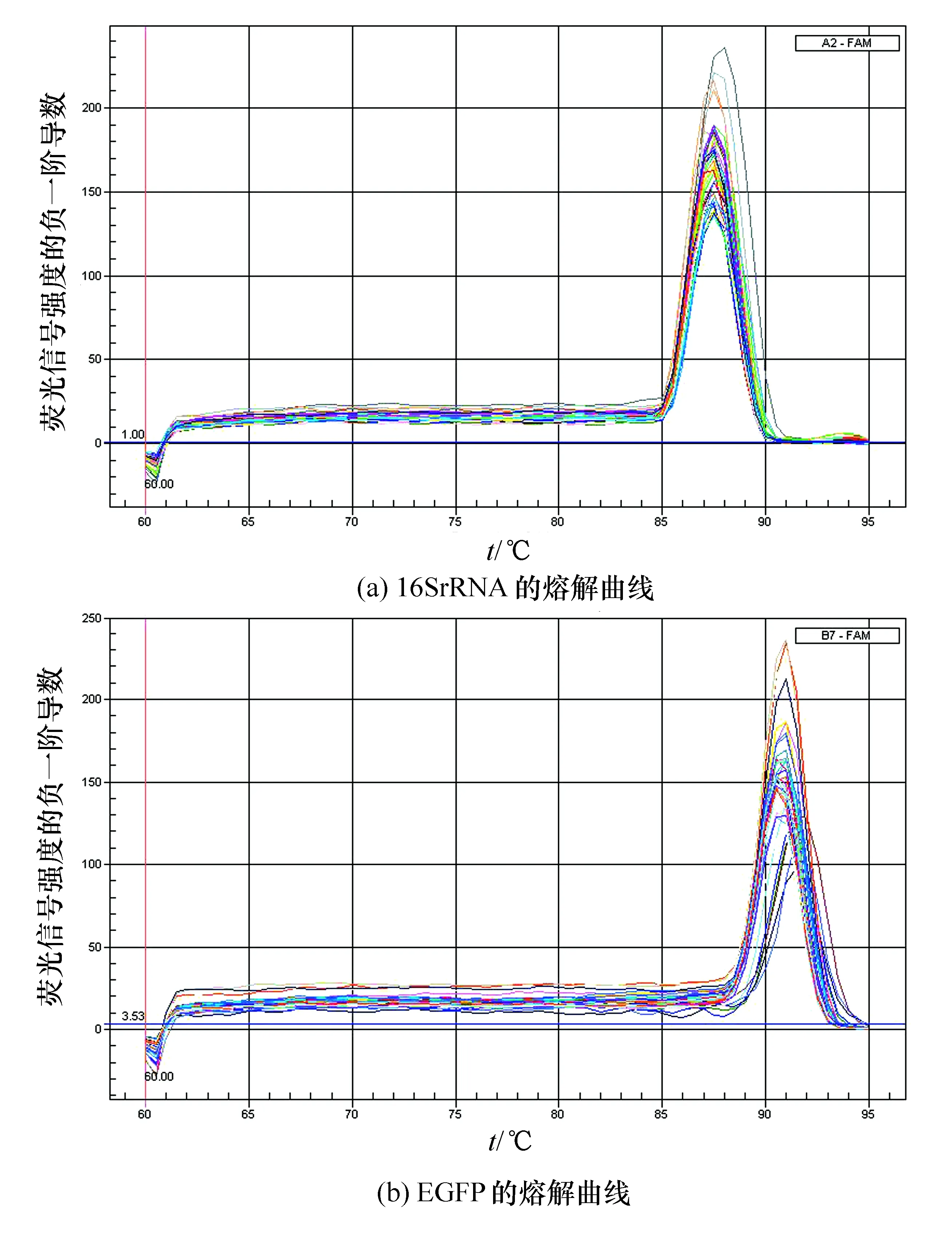
圖2 Real-time PCR熔解曲線
3.4 IPTG最佳誘導條件的確定
采用比較閾值法(2-△△Ct法)對實時熒光定量結果進行分析。由軟件對EGFP基因和16s rRNA基因的擴增曲線進行分析,分別得到相應的Ct(cycle threshold)值,以樣本中的16s rRNA的Ct值作為內參,根據公式2-ΔΔCt= 2-(Ct目的基因-Ct內參)處理組-(Ct目的基因-Ct內參)對照組計算各實驗組的2-ΔΔCt值,將對照組的2-ΔΔCt值設為 1,實驗組
與之相比,進行相對定量。結果見圖3,當IPTG濃度為270 μmol/L、誘導溫度為16 ℃、誘導時間為18 h時,EGFP mRNA的表達量最大。在教學、科研工作中,可以根據實際情況選擇合適的誘導條件:可以選擇低溫(16 ℃)、長時間誘導;也可以選擇高溫(37 ℃)、短時間誘導,節約時間,提高實驗效率。
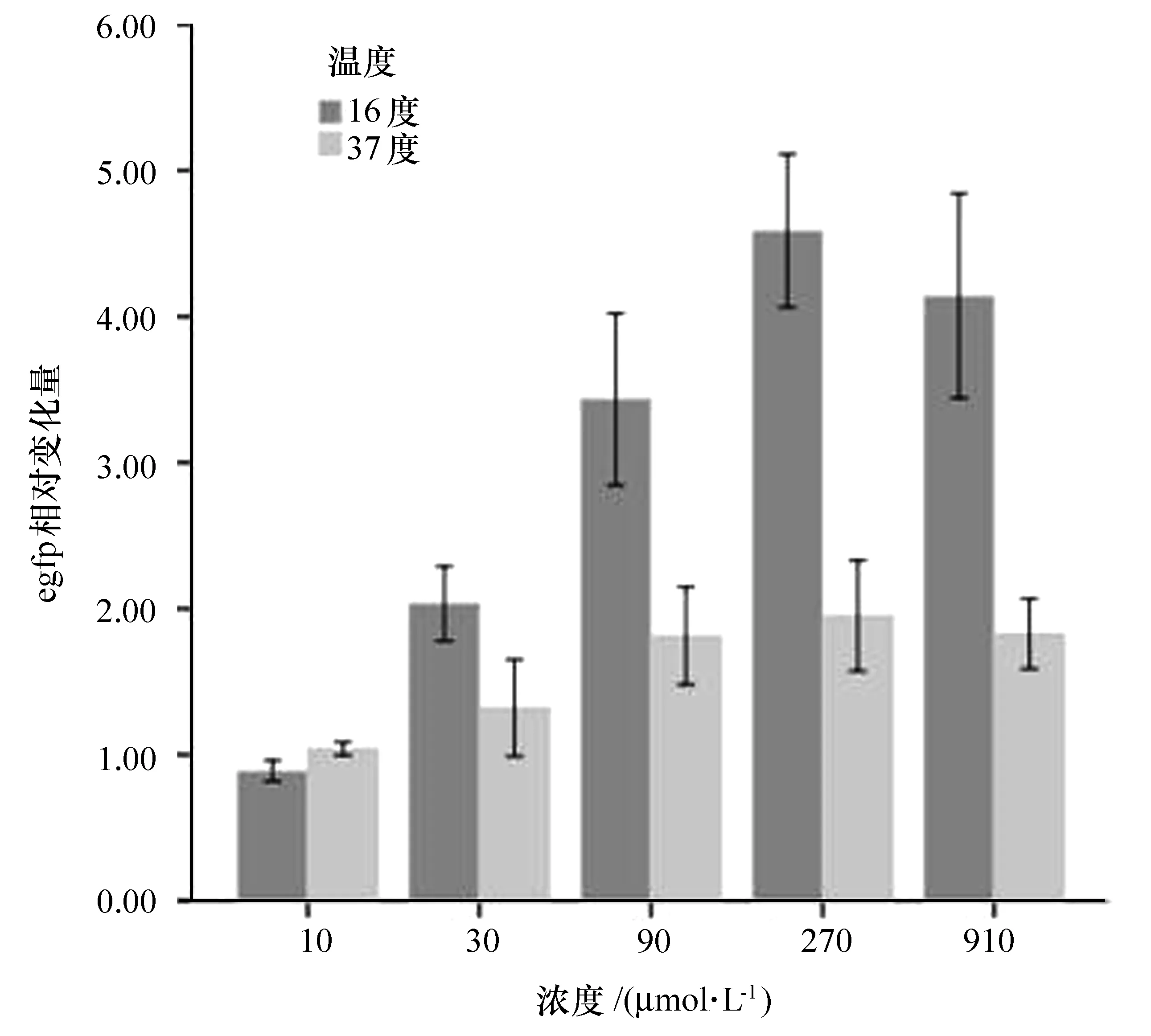
圖3 IPTG不同溫度、不同濃度誘導大腸桿菌中EGFP的表達
4 教學探討
4.1 擴增產物檢測方法的選擇
常用的實時定量PCR擴增產物檢測方法主要有染料法和熒光探針法2種。
(1) 雙鏈DNA結合染料。某些染料能結合到雙鏈DNA上,發射熒光,并且結合后的染料發光強度明顯增加。隨著PCR過程中雙鏈的指數級增長,更多的染料結合到雙鏈DNA中,將信號進一步放大。當DNA變性時,信號又降下來。通過檢測結合到雙鏈DNA上的熒光染料,可以觀察到一個動態的擴增過程。但該方法有2個缺點: 一是它的特異性完全依賴于引物,二是熒光信號的強弱依賴于雙鏈DNA的質量而不是分子數,因而特異性相對較低[5]。
(2) 熒光探針法。熒光探針法主要包括水解探針(hydrolysis probe)、分子信標(molecular beacon)和雜交探針(hybridization probe)等[6-7],利用各種不同探針與DNA特異性的結合,提高檢測的靈敏度,但需要合成高成本的熒光探針,成本較高。
本實驗采用實時熒光定量PCR中常用的SYBR Green Ⅰ染料法。SYBR GreenⅠ是一種非飽和性熒光染料,能夠結合到雙鏈DNA的小溝部位,實時監測擴增進程,而且價格相對較低、通用性好,能夠滿足一般的教學需求。但是,由于SYBR GreenⅠ能與任何ds DNA結合,因此它也能與非特異性的ds DNA(如引物二聚體)結合,使實驗產生假陽性結果[8],影響定量的準確性。為提高檢測結果的靈敏度,本實驗選用大腸桿菌中高度保守的16S rRNA序列設計引物,并確立最佳的退火溫度,結合熔解曲線(melting curve)優化反應條件,排除非特異性擴增和引物二聚體與染料非特異性結合而引起的干擾。
4.2 內參基因的選擇
合適的內參基因對于目的基因的數據校正非常重要,正確選擇內參基因可以降低起始樣本質和量的誤差以及反應效率的誤差[9]。理想的內參基因應滿足以下條件:在分析的組織或樣品中表達穩定;表達不受內源性或外源性因素的影響;表達水平與目的基因表達水平相近;不存在假基因(pseudogene)的干擾。
因此在進行實時熒光定量PCR分析時,應根據實際情況選擇合適的內參基因,以提高實驗數據的準確性和可靠性。
4.3 數據分析
常見的實時熒光定量分析包括相對定量和絕對定量兩種方法。
(1) 相對定量。通過比較目的基因和內參基因的相對含量變化來判斷基因轉錄的變化。
(2) 絕對定量。使用一系列已知起始拷貝數的標準品繪制標準曲線,建立Ct值與起始模板量對數值之間的線性關系。通過未知樣品的Ct值,即可從標準曲線上計算出未知樣品的起始拷貝數。
本實驗采用了相對定量中常用的2-△△Ct法,其中△△Ct=(Ct目的基因-Ct內參)實驗組-(Ct目的基因-Ct內參)對照組。實驗中,要求學生了解實時熒光定量PCR的擴增曲線、熒光閾值、Ct值3個概念,掌握熒光閾值設定的基本原則、明確Ct值的概念以及以Ct 值進行定量的原理。
4.4 實驗注意事項
(1) RNA提取過程中所用的槍頭、離心管等皆為RNase free,且經高壓滅菌處理。
(2) 菌體生長到對數生長期,OD600達到0.6~0.8之間時,再進行誘導培養。
(3) IPTG濃度梯度設計要合理,以保證熒光定量分析時Ct值具有明顯的梯度差,利于數據分析[10]。
(4) 為盡量減少操作誤差,在制備反應體系時應先制備混合反應液,混勻后再分裝到PCR反應管中。
(5) 為提高實驗結果的可靠性,需要設置重復實驗和陰性對照。設置重復性實驗的目的在于按照統計學要求,對數據進行嚴格的校正以消除誤差[8];設置陰性對照的目的在于檢測實驗中所用的引物、模板、酶等是否受到污染,若體系中存在污染,則實驗結果不可用。
(6) 由于總RNA得率不同、RNA反轉錄為cDNA的效率不同等客觀因素,用于定量分析的初始樣品濃度不同。雖然實時定量PCR實驗中會用一些看家基因來校正因樣品初始濃度不同而造成的差異,但是建議保持初始模板量(RNA或cDNA)一致,盡量減少實驗誤差,提高結果的可靠性。
5 結語
由于受操作復雜、實驗周期長、成本高等因素影響,目前在科研實驗室廣泛應用的實時定量PCR分析技術難以應用于本科實驗教學。為此設計“實時熒光定量PCR分析IPTG誘導EGFP基因在大腸桿菌中的異源表達”綜合性實驗,通過培養誘導菌體蛋白表達、提取大腸桿菌總RNA、反轉錄、實時定量PCR、結果分析與討論,使學生有機會在16個學時里就可以學習到這項先進的分析技術,開闊視野,為今后自主開展科學研究工作打下堅實的基礎。
參考文獻(References)
[1] 董小林.昆蟲總RNA提取及實時熒光定量PCR方法探討[J].應用昆蟲學報,2013,50(5):1469-1473.
[2] 靳溪,薛秀花,李潔,等.實時熒光定量PCR技術在發育生物學實驗[J].實驗室研究與探索,2014,33(7):214-217.
[3] 易健明,屈武斌,張成崗.實時熒光定量的數據分析方法[J].生物技術通訊,2015,26(1):140-145.
[4] 楊怡姝,孫曉娜,王小利,等.實時熒光定量PCR 技術的操作實踐[J].實驗室研究與探索,2011,30(7):15-19.
[5] 李小菊,韓際宏,石建黨.分子生物學實驗教程[M].北京:高等教育出版社,2015:83-92.
[6] 王玉倩,薛秀花.實時熒光定量 PCR技術研究進展及其應用[J].生物學通報,2016,51(2):1-4.
[7] 陳旭,齊鳳坤,康立功,等.實時熒光定量 PCR技術研究進展及其應用[J].東北農業大學學報,2010,41(8):148-155.
[8] 薛霜,獨軍政,高閃電,等.實時熒光定量PCR技術研究進展及其在獸醫學中的應用[J].中國農學通報,2010,26(7):11-15.
[9] 紀東,辛紹杰.實時熒光定量PCR的發展和數據分析[J].生物技術通訊,2009,20(4):598-600.
[10] 湯海峰,姜丹,孟令軍,等.熒光定量法分析液體發酵中雜菌污染的實驗設計[J].實驗技術與管理,2016,33(6):50-54.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
