Keggin型多酸負(fù)載的單原子催化劑(M1/POM, M = Ni, Pd, Pt, Cu,Ag, Au, POM = [PW12O40]3-)活化氮?dú)夥肿拥拿芏确汉碚撚?jì)算研究
2018-04-10 11:24:17尹玥琪蔣夢(mèng)緒劉春光
物理化學(xué)學(xué)報(bào) 2018年3期
尹玥琪,蔣夢(mèng)緒,劉春光
1 引言
氮元素是自然界中十分重要的元素之一,它是植物生長(zhǎng),人類(lèi)生產(chǎn)、生活必不可少的基本元素。由于氮元素在自然界中主要以氮?dú)夥肿拥男问酱嬖冢獨(dú)獾幕瘜W(xué)性質(zhì)又十分穩(wěn)定,因此不能被動(dòng)植物直接吸收和利用。自然界中主要依靠閃電,或者植物中的固氮菌來(lái)實(shí)現(xiàn)氮元素的轉(zhuǎn)化1,2。隨著工、農(nóng)業(yè)生產(chǎn)對(duì)氮元素需求量的不斷增加,僅僅依靠生物固氮已經(jīng)遠(yuǎn)遠(yuǎn)不能滿足現(xiàn)代社會(huì)發(fā)展的需要。因此,常溫、常壓下人工固氮的方法被各界學(xué)者廣泛關(guān)注。采用化學(xué)方法,制備出類(lèi)似植物“固氮菌”的物質(zhì),使空氣中的氮在常溫常壓下就能轉(zhuǎn)變?yōu)殇@態(tài)氮或氨態(tài)氮供人們利用,成為學(xué)界關(guān)注的前沿?zé)狳c(diǎn)問(wèn)題之一3-8,特別是雙氮配位化學(xué),由于在人工固氮方面的潛在應(yīng)用而特別受到關(guān)注9,10。在過(guò)去的半個(gè)多世紀(jì)里,許多學(xué)者認(rèn)為具有潛在固氮作用的過(guò)渡金屬-氮?dú)馀浜衔锞哂谢罨獨(dú)夥肿拥哪芰?1-13。目前已經(jīng)報(bào)道了許多具有新穎化學(xué)結(jié)構(gòu)的過(guò)渡金屬-氮?dú)馀浜衔?4,15。其中Chatt循環(huán)是這一領(lǐng)域最具代表性的范例之一(圖1)。該循環(huán)基于一個(gè)單核鉬-氮?dú)忪⑴浜衔铮ㄟ^(guò)連續(xù)的多步質(zhì)子耦合電子轉(zhuǎn)移,最終將一個(gè)配位的氮?dú)夥肿愚D(zhuǎn)化為兩個(gè)氨氣分子,而單核鉬膦配合物在循環(huán)過(guò)程中起到了催化劑的作用。
充分應(yīng)用負(fù)載型金屬催化劑每一個(gè)原子的最有效的方法是縮小金屬體相材料的尺寸到單個(gè)的獨(dú)立原子,含有孤立的單原子作為活性中心的負(fù)載型金屬催化劑被定義為單原子催化劑(SACs)。由于SACs是由孤立的單金屬原子分散在負(fù)載材料表面組成,因此,從理論上說(shuō),分散度應(yīng)該是100%。這樣的催化劑與傳統(tǒng)的負(fù)載型金屬納米粒子催化劑是截然不同的。在過(guò)去的幾年里,有很多例子毋庸置疑的證明了負(fù)載型SACs優(yōu)異的催化行為。SACs不但表現(xiàn)出催化活性而且具有最高的催化活性,并且在催化反應(yīng)過(guò)程中十分的穩(wěn)定。這主要是由于金屬單原子與載體表面錨定位點(diǎn)之間具有較強(qiáng)的鍵連作用16-19。例如:對(duì)于CO的氧化和優(yōu)先氧化,Pt1/FeOx單原子催化劑的活性是亞納米尺寸催化劑活性的2-3倍,并且,在較長(zhǎng)的檢測(cè)周期內(nèi)都具有很高的穩(wěn)定性16。對(duì)于3-硝基苯乙烯的加氫反應(yīng),Pt1/FeOx單原子催化劑的周轉(zhuǎn)頻率達(dá)到了1500 s-1,是目前報(bào)道的最好結(jié)果的20倍左右20。另外,對(duì)于氨基苯乙烯的加氫反應(yīng),該催化劑的選擇性達(dá)到了99%,這是目前Pt族催化劑獲得的最大值。將Pd單原子分散到乙烯醇鹽的表面,乙烯醇鹽被穩(wěn)定在超薄的TiO2納米片上。通過(guò)此方法合成的Pd1/TiO2單原子催化劑在C=C鍵加氫反應(yīng)中表現(xiàn)出了很高的催化活性,超過(guò)市售Pd催化劑表面Pd原子的九倍。乙醛加氫催化反應(yīng)增加了55倍21。孤立的單金屬原子分散在恰當(dāng)?shù)妮d體表面表現(xiàn)出的優(yōu)異的催化活性打開(kāi)了通往多相催化前沿的大門(mén)。
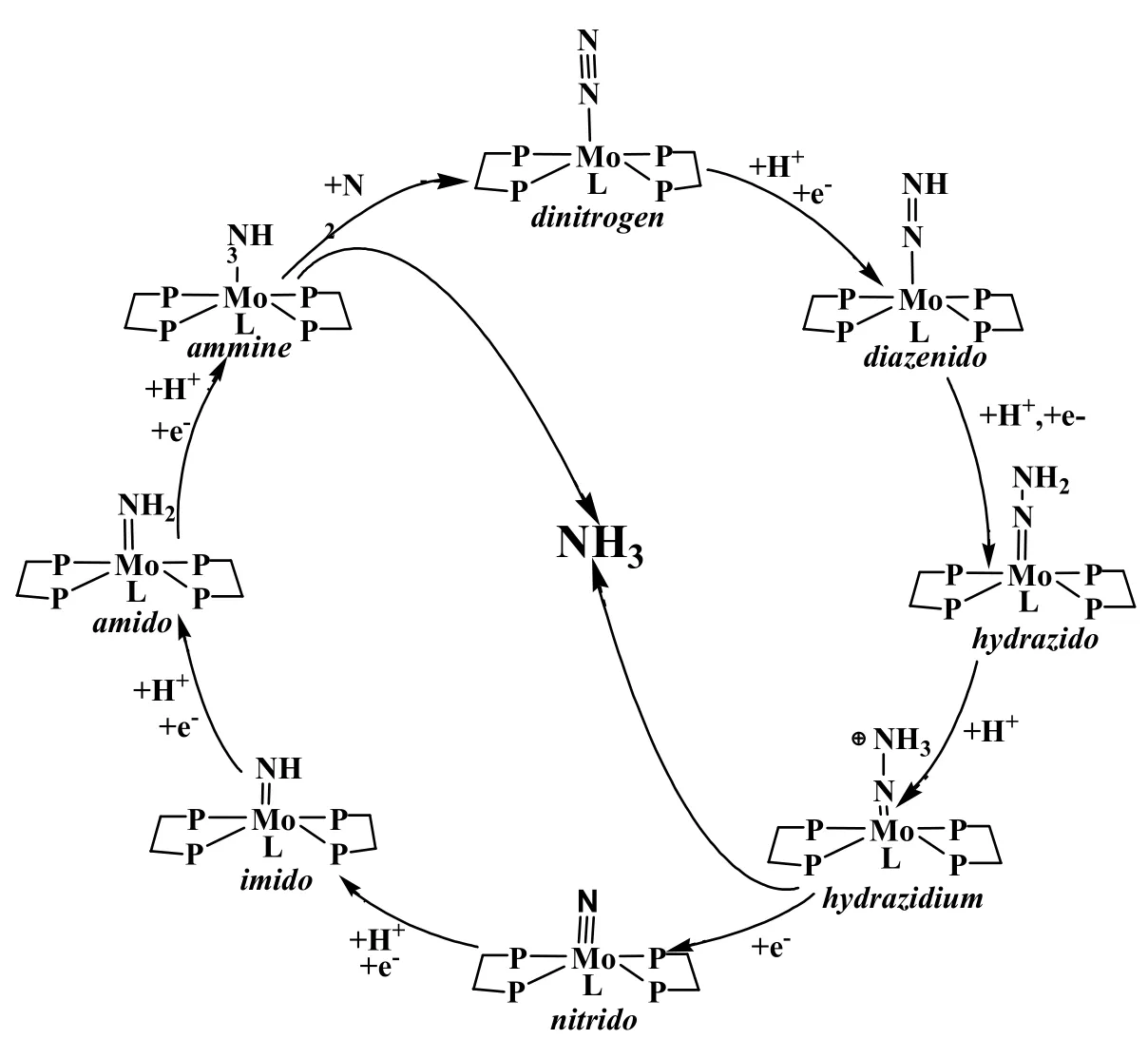
圖1 單核鉬膦配合物的固氮Chatt循環(huán)Fig.1 The chatt cycle for nitrogen fixation on a mononuclear molybdenum-phosphine complex.
多酸(POMs)全稱(chēng)多金屬氧酸鹽,是一類(lèi)由氧原子橋接金屬原子形成的金屬-氧簇化合物,由于它具有超強(qiáng)的酸性和優(yōu)良的氧化還原性,成為一種新型的兼具酸和氧化還原催化活性的雙功能催化劑。POMs類(lèi)化合物催化活性高,選擇性好,對(duì)設(shè)備腐蝕性小,同時(shí)又不產(chǎn)生污染,是一種環(huán)境友好和很有發(fā)展前途的綠色催化劑,目前已經(jīng)被廣泛應(yīng)用于酯類(lèi)水解、烯烴環(huán)氧化、烷烴羥基化,光催化分解水等一些意義重大的反應(yīng)中22-30。不同于金屬氧化物,POMs的體相材料由孤立的陰離子基團(tuán)與抗衡離子構(gòu)成,而表現(xiàn)為不連續(xù)的結(jié)構(gòu)。更形象的說(shuō),POMs類(lèi)化合物更像是由獨(dú)立的金屬氧化物“碎片”組成。早在1970年代,這類(lèi)化合物就被選擇作為催化劑載體材料31。最近,以Keggin型POM為載體的SACs(Pt1/POM,POM =[PW12O40]3-),已經(jīng)被成功合成32,33,并且Pt1/POM催化劑在芳烴加氫反應(yīng)中表現(xiàn)出了優(yōu)異的催化活性和選擇性32。
本文采用密度泛函理論(DFT)計(jì)算方法探究了一系列以多酸為載體的 SACs (M1/POM (M =Ni, Pd, Pt, Cu, Ag, Au, POM = [PW12O40]3-)潛在的活化氮?dú)夥肿拥哪芰Α1疚牡闹饕康脑谟谔接懷芯矿w系的電子結(jié)構(gòu)和金屬-氮?dú)獬涉I相互作用與活化氮?dú)庵g的內(nèi)在聯(lián)系,為后續(xù)的氮轉(zhuǎn)移反應(yīng)做好前期理論工作。
2 計(jì)算細(xì)節(jié)
所有的分子幾何構(gòu)型都采用 meta-廣義梯度近似(meta-GGA)的交換相關(guān)泛函 M06L進(jìn)行優(yōu)化。M06L泛函采用一種平衡的方法將自旋能量密度分配到交換和相關(guān)泛函中。前期的理論研究工作已經(jīng)證明 M06L泛函在計(jì)算由主族和過(guò)渡金屬元素組成化合物的熱化學(xué)、動(dòng)力學(xué)、非共價(jià)鍵相互作用和頻率等都表現(xiàn)出了較好的行為34。同時(shí),理論計(jì)算表明 M06L泛函在預(yù)測(cè)多酸化合物的分子幾何結(jié)構(gòu)、振動(dòng)頻率和熱化學(xué)等性質(zhì)都表現(xiàn)出了較高的準(zhǔn)確性30。主族元素采用雙ζ層劈裂價(jià)基加極化函數(shù)6-31G(d)來(lái)構(gòu)筑分子的哈密頓量,考慮到相對(duì)論效應(yīng),金屬元素采用LANL2DZ基組35,36。為了確定計(jì)算的分子結(jié)構(gòu)是勢(shì)能面上的極小值點(diǎn)和獲得熱力學(xué)性質(zhì),在相同的理論水平上進(jìn)行了頻率計(jì)算。溶劑化效應(yīng)通過(guò)采用SCRF(自洽場(chǎng)反應(yīng))方法的 IEFPCM(積分方程形式的連續(xù)極化模型)進(jìn)行計(jì)算37。基于優(yōu)化的分子幾何結(jié)構(gòu),采用自然鍵軌道理論分析(NBO)38程序檢驗(yàn)所研究化合物的電子結(jié)構(gòu)。所有的計(jì)算都是通過(guò)Gaussian 09程序包完成39。
N2的吸附能計(jì)算公式為:
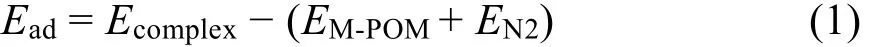
Εcomplex是金屬-氮?dú)?POM 配合物的能量,ΕM-POM是過(guò)渡金屬-氮?dú)馀浜衔锏哪芰浚2是自由氮?dú)夥肿拥哪芰俊?/p>
M1/POM 的過(guò)渡金屬與多酸載體之間的吸附能計(jì)算公式為:
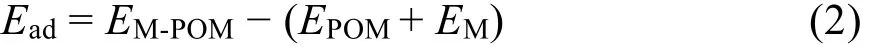
ΕM-POM是POM的能量,ΕPOM是POM的能量,ΕM是游離金屬原子的能量。
3 結(jié)果與討論
過(guò)渡金屬原子往往會(huì)依據(jù)不同的配位環(huán)境而表現(xiàn)出不同的自旋態(tài)。根據(jù)實(shí)驗(yàn)和理論計(jì)算表明,在以多酸為載體的M1/POM體系中孤立的金屬原子往往錨定在多酸表面橋氧原子組成的四重洞位上(見(jiàn)圖2)40。我們采用DFT-M06L計(jì)算了本文中每個(gè)體系所有可能自旋態(tài)的總能量(總能量包括電子能量和零點(diǎn)校正能量),計(jì)算結(jié)果列于表S1(見(jiàn)Supporting Information)中。對(duì)于處于同一族的Ni、Pd、Pt三種金屬原子,它們都具有d8電子組態(tài),在原子的5個(gè)d軌道上有兩種排布形式,因此,每個(gè)體系都可能具有單重和三重兩個(gè)自旋態(tài)。對(duì)于處于同一族的 Cu、Ag、Au三種金屬原子具有d9電子組態(tài),在原子的5個(gè)d軌道上只有一種排布形式,所以,每個(gè)體系只能具有一個(gè)二重態(tài)。我們的計(jì)算結(jié)果表明前過(guò)渡金屬 Ni1/POM體系具有一個(gè)高自旋的三重基態(tài),后過(guò)渡金屬Pd1/POM和Pt1/POM體系具有低自旋的單重基態(tài)。眾所周知,前過(guò)渡金屬原子往往以高自旋態(tài)最穩(wěn)定30。相反,后過(guò)渡金屬原子主要以低自旋態(tài)能量最低。我們的DFT-M06L計(jì)算結(jié)果和這一結(jié)論是完全一致的。
錨定于多酸表面四重洞位的獨(dú)立金屬原子與配位的氧原子構(gòu)成了一個(gè)四棱錐的幾何結(jié)構(gòu),金屬原子位于四棱錐的頂點(diǎn)上(見(jiàn)圖 2)。通過(guò)DFT-M06L計(jì)算得到的M1/POM體系中金屬原子與四個(gè)配位的氧原子之間的平均距離avM―O列于圖2中。計(jì)算結(jié)果表明avM―O值按照Ni1/POM <Cu1/POM < Ag1/POM < Pd1/POM < Au1/POM <Pt1/POM 的順序遞增。Ni1/POM 體系給出了小的avM-O值。采用M06L計(jì)算獲得的單金屬原子在多酸表面的吸附能(單位均為 kJ·mol-1)按照Ni1/POM (-448.02)< Pt1/POM (-133.34)<Cu1/POM (-116.06)< Pd1/POM (-111.29)<Ag1/POM (-111.04) < Au1/POM (-108.78)順序升高,Ni1/POM 體系給出了最負(fù)的吸附能。大量的實(shí)驗(yàn)研究表明負(fù)載型金屬催化劑中金屬-載體之間相互作用不宜過(guò)強(qiáng)。金屬-載體之間相互作用過(guò)強(qiáng)勢(shì)必會(huì)消弱中心金屬原子與反應(yīng)物分子之間的相互作用,從而降低其催化活性。對(duì)于本文研究的體系,金屬-載體之間相互作用最強(qiáng)的Ni1/POM多酸體系的催化活性將較弱。
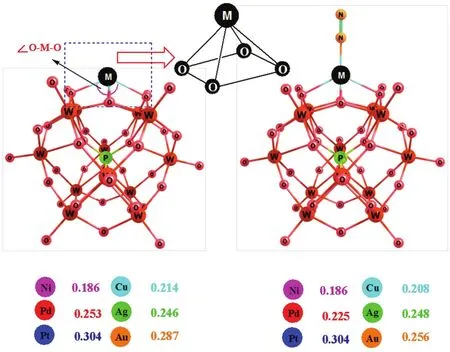
圖2 M1/POM體系吸附氮?dú)馇昂蟮姆肿訋缀谓Y(jié)構(gòu)和金屬與四個(gè)配體氧的平均鍵長(zhǎng)(avM-O, nm)Fig.2 Molecular structure of M1/POM and nitrogen-metal complex and average bond length (avM-O, nm).
值得注意的是Pt1/POM體系也具有較負(fù)的吸附能。這主要由于是Pt1/POM的結(jié)構(gòu)中錨定于多酸表面四重洞位的Pt原子與配位的氧原子構(gòu)成的四棱錐結(jié)構(gòu)并非是正四棱錐,Pt原子向其中的一個(gè)氧原子偏移,這一現(xiàn)象是導(dǎo)致Pt1/POM有較大avM-O值的同時(shí)還具有較負(fù)吸附能的原因,這一特殊結(jié)構(gòu)直接導(dǎo)致它具有較強(qiáng)的活化氮?dú)獾哪芰Α?/p>
為了驗(yàn)證對(duì)這些分子幾何結(jié)構(gòu)上的猜測(cè),對(duì)氮?dú)夥肿釉赟ACs上的吸附能進(jìn)行計(jì)算(金屬原子采用LANL2DZ基組),吸附前后的結(jié)構(gòu)模型在圖2中比較列出。計(jì)算出的數(shù)據(jù)也都列在表1中,可發(fā)現(xiàn)吸附能按照 Pt1/POM < Pd1/POM< Cu1/POM <Ag1/POM < Au1/POM < Ni1/POM 的順序遞增。尤其是Pt1/POM吸附能最負(fù),達(dá)到-239.16 kJ·mol-1,這個(gè)結(jié)果與之前分子幾何結(jié)構(gòu)的預(yù)測(cè)完全一致。
為了分析POM載體在SAC體系中的作用,分別計(jì)算了氮?dú)夥肿釉赑t1/POM、孤立的Pt原子和兩種由八個(gè) Pt原子構(gòu)成的團(tuán)簇(Pt8)上的吸附能。相關(guān)結(jié)果列于表S2中。計(jì)算結(jié)果表明,以多酸為載體 Pt1/POM體系 N2吸附能與獨(dú)立金屬 Pt原子相當(dāng)。但是,比上述兩種團(tuán)簇更負(fù)。這說(shuō)明孤立的Pt原子相比于Pt原子團(tuán)簇有著更加優(yōu)異的氮?dú)馕教匦裕琍OM 載體沒(méi)有影響到獨(dú)立的 Pt原子對(duì)氮?dú)夥肿拥奈叫阅堋?/p>
吸附氮?dú)馇昂蠓肿訋缀谓Y(jié)構(gòu)和相關(guān)參數(shù)比較于圖2中,計(jì)算結(jié)果表明,由于無(wú)機(jī)POM籠的剛性結(jié)構(gòu),在吸附氮?dú)馇昂螅琍OM載體部分幾何結(jié)構(gòu)參數(shù)基本沒(méi)有發(fā)生明顯改變。氮?dú)馕脚浜衔锏年P(guān)鍵分子幾何結(jié)構(gòu)參數(shù)列于表 1中。表中數(shù)據(jù)表明,M―N 鍵長(zhǎng)按照 Pt < Cu < Pd < Au < Ag < Ni的順序遞增,優(yōu)化后 N≡N鍵距按照 Ni < Ag < Cu <Au < Pd < Pt的順序遞增。為了進(jìn)一步探究負(fù)載金屬單原子與氮?dú)庵g的相互作用,對(duì)研究體系的電子結(jié)構(gòu)進(jìn)行了NBO分析,計(jì)算所得數(shù)據(jù)也列在表 1中,M―N鍵的 WBI (Wiberg)鍵序按照Pt(0.916) > Pd(0.631) > Cu(0.518) > Au(0.481) >Ag(0.262) > Ni(0.005)的順序遞減,N≡N 之間的WBI鍵序按照 Ni(2.970) > Ag(2.825) > Cu(2.760) >Au(2.704) > Pd(2.648) > Pt(2.528)的順序遞減。化學(xué)鍵鍵長(zhǎng)與成鍵原子半徑和成鍵原子之間的距離有關(guān),WBI鍵序可以剔除原子半徑因素對(duì)鍵長(zhǎng)的影響,是描述分子中相鄰原子之間的成鍵強(qiáng)度的物理量。上述計(jì)算結(jié)果表明,Pt1/POM 給出了最大的WBI(M―N)鍵序,和最小的WBI(N≡N)鍵序,說(shuō)明了明顯的活化氮?dú)夥肿拥哪芰Α_@一結(jié)果為Pt1/POM 體系潛在的固定氮?dú)夥肿硬⑹蛊浠罨峁┝烁辛Φ淖C據(jù)。另外對(duì)于負(fù)載過(guò)渡金屬Ni的體系來(lái)說(shuō),其 WBI(M―N)值僅有 0.005,說(shuō)明過(guò)渡金屬與氮?dú)庵g的相互作用極弱,金屬Ni與四個(gè)氧配體之間的WBI鍵序都達(dá)到了0.4左右,說(shuō)明Ni與多酸之間相互作用較強(qiáng),反而與氮?dú)庵g的相互作用很小,甚至不能達(dá)到使氮?dú)夤潭ㄔ诙嗨岽呋瘎┥系哪康模虼薔i1/POM體系不能達(dá)到活化氮?dú)獾哪康摹1?中給出研究體系的NBO電荷,由表中數(shù)據(jù)可知吸附的氮?dú)夥肿硬糠值腘BO電荷遞增順序?yàn)椋篜d(-0.209) < Pt(-0.197) <Au(-0.121)< Ag(-0.089)< Cu(-0.083),可以看出,這些吸附后的氮?dú)夥肿与姾啥际秦?fù)的。和中性的自由氮分子相比,氮?dú)馍系牟糠重?fù)電荷表明發(fā)生了電荷轉(zhuǎn)移。Pt1/POM、Pd1/POM體系中氮?dú)獠糠謳в械呢?fù)電荷達(dá)到~0.2e,表明了相對(duì)明顯的電荷轉(zhuǎn)移。
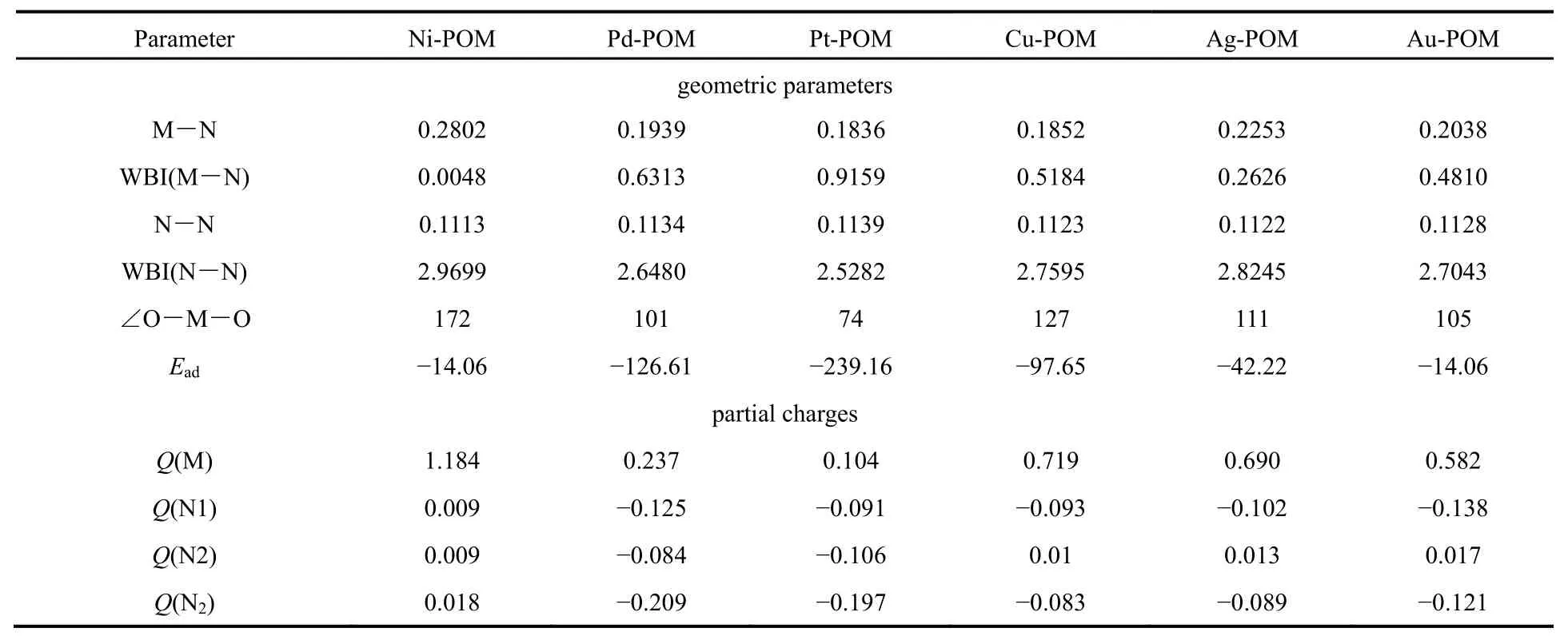
表1 DFT-M06L計(jì)算獲得的氮?dú)馕侥?Ead, kJ·mol-1)、M1/POM (M = Ni, Pd, Pt, Cu, Ag, Au)體系的鍵長(zhǎng)(nm),金屬與兩個(gè)配體氧之間的鍵角(∠O-M-O, °)、WBI鍵序(WBI)和NBO電荷(Q, a.u.)Table 1 DFT-M06L-derived adsorption energy (Ead, kJ·mol-1), bond length (nm), angle of metal atom and two oxygen atoms (∠O-M-O, °), WBI bond order (WBI) and NBO partial charge (Q, a.u.) in M1/POM (M = Ni, Pd, Pt, Cu, Ag, Au).
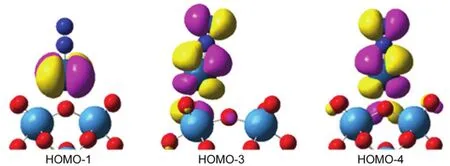
圖3 Pt1/POM的前線分子軌道分布圖Fig.3 Frontier molecular orbitals of the Pt-nitrogen POM complex.
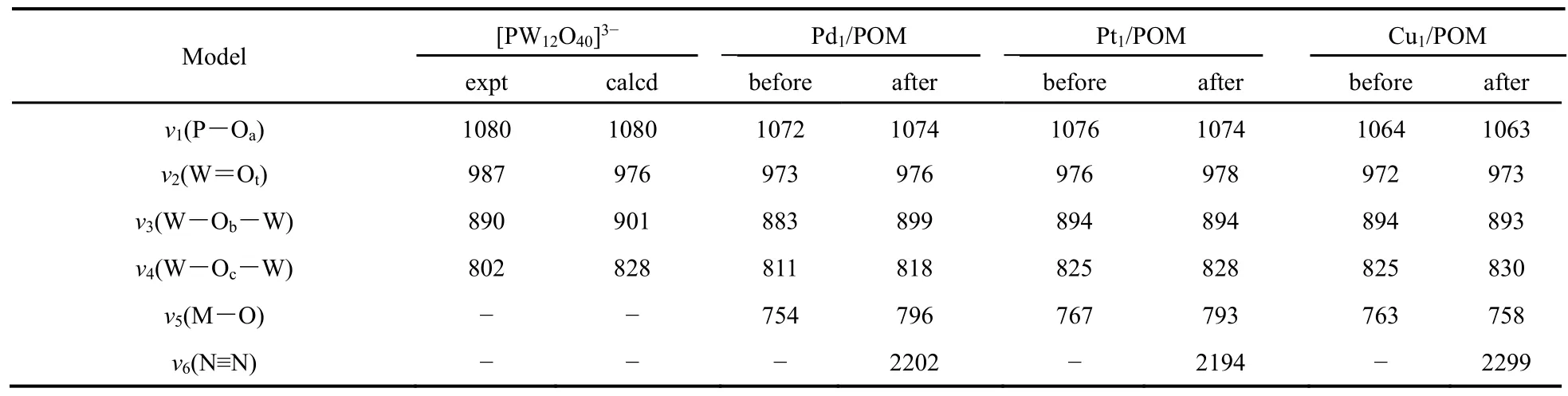
表2 M1/POM催化體系吸附氮?dú)馇昂蠹t外光譜特征峰振動(dòng)頻率(單位:cm-1)Table 2 Calculated and experimental vibrational frequencies (in cm-1) and the assigned bands of the series of POM complexes studied here.
前線分子軌道理論可以解釋成鍵規(guī)律的定性理論。通過(guò)上文的NBO分析和成鍵性質(zhì)分析可知Pt1/POM 體系相對(duì)于其它體系來(lái)說(shuō)有著較為明顯的活化氮?dú)夥肿拥哪芰Γ虼讼挛闹兄饕訮t1/POM體系為例分析它們的前線分子軌道特征。Pt1/POM體系前線分子軌道如圖 3所示,可以看到,HOMO - 1軌道主要分布在負(fù)載的Pt原子上,是Pt原子的dxy軌道,表現(xiàn)為非鍵道。HOMO - 3和HOMO - 4這兩個(gè)軌道是由氮?dú)夥肿拥摩?反鍵軌道和過(guò)渡金屬Pt原子對(duì)稱(chēng)匹配的dxz和dyz軌道上的電子云重疊構(gòu)成的Pt-N2成鍵軌道。如上文所述,NBO電荷分析證明 Pt-N2成鍵過(guò)程發(fā)生的金屬到氮?dú)夥肿拥碾姾赊D(zhuǎn)移。根據(jù) HOMO - 3和HOMO - 4軌道分析表明,Pt金屬原子到氮?dú)夥肿由系碾姾赊D(zhuǎn)移,主要是由Pt金屬原子的dxz、dyz軌道上的電子填充到了氮?dú)獾?π*反鍵軌道上引起。填入π*反鍵軌道的電子增長(zhǎng)了N≡N鍵之間的距離,有效的活化了氮?dú)夥肿印R陨戏治隹梢酝ㄟ^(guò)d2xyπ2xzπ2yz電子組態(tài)進(jìn)行充分概括。
計(jì)算的M1/POM體系的紅外光譜列于表2中。眾所周知,完整的Keggin型POM是一個(gè)由氧原子連接鎢原子形成的籠狀結(jié)構(gòu),內(nèi)部包含四面體磷酸基團(tuán),整個(gè)結(jié)構(gòu)具有Td對(duì)稱(chēng)。實(shí)驗(yàn)上所測(cè)得的紅外光譜數(shù)據(jù)表明,完整Keggin型POM具有四個(gè)明顯的特征譜帶(P-Oa,W=Ot,W-Ob-W和W-Oc-W鍵):P-Oa鍵的不對(duì)稱(chēng)伸縮振動(dòng)出現(xiàn)在1080 cm-1處(Oa是四面體磷酸基團(tuán)中的氧原子);W=Ot鍵的不對(duì)稱(chēng)伸縮振動(dòng)出現(xiàn)在987 cm-1處(Ot是端氧原子);W-Ob-W 鍵不對(duì)稱(chēng)伸縮振動(dòng)出現(xiàn)在890 cm-1(Ob代表橋氧原子);W-Oc-W 鍵的不對(duì)稱(chēng)伸縮振動(dòng)出現(xiàn)在 802 cm-1的是(Oc代表處于轉(zhuǎn)角位置的橋氧原子)30。為了驗(yàn)證我們計(jì)算方法的可靠性,首先采用相同的理論水平計(jì)算了完整的Keggin型POM,[PW12O40]3-,的紅外光譜,計(jì)算結(jié)果也列于表2中,P-Oa(1080 cm-1)、W=Ot(976 cm-1),W-Ob-W (901 cm-1)和 WOc-W (828 cm-1),與實(shí)驗(yàn)測(cè)得的頻率相一致(見(jiàn)表2),證明了計(jì)算方法的可行性。
當(dāng)多酸負(fù)載過(guò)渡金屬原子后,完整的 Keggin型 POM 四個(gè)明顯的特征吸收峰基本沒(méi)有發(fā)生變化,但是在750-770 cm-1范圍內(nèi)出現(xiàn)了新的吸收峰。如表2所示,Pd1/POM、Pt1/POM、Cu1/POM的新的特征峰分別出現(xiàn)在754、767、763 cm-1的位置處。對(duì)這些峰的振動(dòng)模式分析表明主要是由W-Oc-W 鍵的伸縮振動(dòng)引起。如前文所述,負(fù)載的單金屬原子錨定于Keggin型POM表面的四重洞位上,主要表現(xiàn)為金屬原子與W-Oc-W鍵中的Oc原子發(fā)生直接相互作用。這種成鍵方式影響了W-Oc-W鍵的伸縮振動(dòng)。因此可以判斷新出現(xiàn)的特征峰是由于負(fù)載單金屬原子后從完整的Keggin型POM的W-Oc-W特征峰中分離出來(lái)的。
吸附氮?dú)夂笮纬傻呐浜衔锏募t外光譜數(shù)據(jù)也列在表2中。值得注意的是,吸附氮?dú)夥肿又螅?(M-O)發(fā)生了移動(dòng),這可能是由于金屬與氮?dú)庀嗷プ饔脤?dǎo)致金屬與多酸表面四重洞位上的氧原子之間的相互作用減弱引起的。吸附氮?dú)夥肿雍螅诩t外光譜上出現(xiàn)了另一個(gè)明顯的紅外特征吸收峰,對(duì)應(yīng)于氮?dú)夥肿覰≡N鍵伸縮振動(dòng)。自由氮?dú)獾募t外特征吸收峰出現(xiàn)在2331 cm-1,N≡N鍵長(zhǎng)為0.1098 nm。對(duì)雙氮分子的紅外光譜研究表明,當(dāng)?shù)獨(dú)夥肿拥腘≡N鍵伸長(zhǎng)時(shí),N≡N鍵伸縮振動(dòng)頻率將會(huì)減小。例如偶氮苯的紅外吸收峰出現(xiàn)在1442 cm-1,N≡N鍵長(zhǎng)為0.1255 nm;肼的紅外吸收峰出現(xiàn)在1111 cm-1,N≡N鍵長(zhǎng)為0.1460 nm。如上文所述,氮?dú)夥肿拥腘≡N鍵長(zhǎng)的順序?yàn)椋篜t(0.1139 nm) > Pd(0.1134 nm) > Cu(0.1123 nm),計(jì)算出紅外光譜中N≡N鍵伸縮振動(dòng)頻率按照Pt(2194 cm-1) <Pd(2202 cm-1) < Cu(2299 cm-1)的順序遞增,與上述規(guī)律完全一致。
4 結(jié)論
采用DFT計(jì)算研究了一系列以POM為載體的SACs(M1/POM;M = Ni, Pd, Pt, Cu, Ag, Au)的分子幾何構(gòu)型、電子結(jié)構(gòu)、紅外光譜。計(jì)算結(jié)果表明這些催化劑具有潛在的活化N2分子的能力,特別是Pt1/POM體系吸附的N2分子給出了最大的N≡N鍵長(zhǎng),同時(shí)NBO分析表明Pt1/POM體系中的WBI(N≡N)的值也最小。因此,在六種研究體系中Pt1/POM的活化N2效果較好。前線分子軌道分析發(fā)現(xiàn)Pt1/POM中Pt金屬原子的dxz、dyz軌道與氮?dú)獾摩?反鍵軌道相互重疊使多酸載體與N2之間成鍵,并且金屬原子中的部分電子轉(zhuǎn)移到了N2的π*反鍵軌道導(dǎo)致N≡N鍵增長(zhǎng)。通過(guò)對(duì)M1/POM體系的紅外光譜的DFT計(jì)算結(jié)果與實(shí)驗(yàn)數(shù)據(jù)相比較發(fā)現(xiàn),本文采用的理論方法很好的重現(xiàn)了過(guò)渡金屬取代Keggin結(jié)構(gòu)的雜多酸的紅外光譜,對(duì)Pt1/POM,Pd1/POM和Cu1/POM的特征吸收峰分析表明,由于引入單原子到多酸表面產(chǎn)生了新的特征吸收峰。
Supporting lnformation:available free of charge νia the internet at http://www.whxb.pku.edu.cn.
(1)Burris, R. H. J. Biol. Chem. 1991, 266, 9339.
(2)Shah, V. K.; Brill, W. J. Proc. Νatl. Acad. Sci. U. S. A. 1977, 74,3249. doi: 10.1073/pnas.0507853103
(3)Chakrabarti, P.; Woo, D.;Kornuc, J. J.; Rees, D. C. Science 1992,257, 1653. doi: 10.1126/science.1529353
(4)Kirn, J.; Rees, D. C. Νature 1992, 360, 553. doi: 10.1038/360553a0
(5)Kirn, J.; Rees, D. C. Science 1992, 257, 1677.doi: 10.1126/science.1529354
(6)Chan, M. K.; Kirn, J.; Rees, D. C. Science 1993, 260, 792.doi: 10.1126/science.8484118
(7)Kirn, J.; Woo, D.; Rees, D. C. Biochemistry 1993, 32, 7104.doi: 10.1021/bi00079a006
(8)Kirn, J.; Rees, D. C. Biochemistry 1994, 33, 389.doi: 10.1021/bi00168a001
(9)Jia, H. P.; Quadrelli, E. A. Chem. Soc. Reν. 2014, 43, 547.doi: 10.1039/c3cs60206k
(10)MacKay, B. A.; Fryzuk, M. D. Chem. Reν. 2004, 104, 385.doi: 10.1021/cr020610c
(11)Yandulov, D. V.; Schrock, R. R. Science 2003, 301, 76.doi: 10.1126/science.1085326
(12)Arashiba, K.; Miyake, Y.; Nishibayashi, Y. Νat. Chem. 2011,3, 120. doi: 10.1038/nchem.906
(13)Anderson, J. S.; Rittle, J.; Peters, J. C. Νature 2013, 501, 84.doi: 10.1038/nature12435
(14)Allen, A. D.; Harris, R. O.; Loescher, B. R.; Stevens, J. R.;Whiteley, R. N. Chem. Reν. 1973, 73, 11.doi: 10.1021/cr60281a002
(15)Hoffman, B. M.; Lukoyanov, D.; Yang, Z. Y.; Dean, D. R.;Seefeldt, L. C. Chem. Reν. 2014, 114, 4041.doi: 10.1021/cr400641x
(16)Qiao, B.; Wang, A.; Yang, X.; Allard, L. F.; Jiang, Z.; Cui,Y.; Liu, J.; Li, J.; Zhang, T. Νat. Chem. 2011, 3, 634.doi: 10.1038/nchem.1095
(17)Yang, X. F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T.Acc. Chem. Res. 2013, 46, 1740.doi: 10.1021/ar300361m.Epub 2013 Jul1
(20)Wei, H.; Liu, X. Y.; Wang, A.; Zhang, L.; Qiao, B. T.; Yang,Y. F.; Huang, Y. Q.; Miao, S.; Liu, J.; Zhang, T. Νat.Commun. 2013, 5, 5634. doi: 10.1038/ ncomms6634
(21)Liu, P.; Zhao, Y.; Qin, R.; Mo, S.; Chen, G.; Gu, L.;Chevrier, D. M.; Zhang, P.; Guo, Q.; Zang, D.; Wu, B.; Fu,G.; Zheng, N. Science 2016, 352, 797.doi: 10.1126/science.aaf5251
(22)Katsoulis, D. E. Chem. Reν. 1998, 98, 359.doi: 10.1021/cr960398a
(23)Dolbecq, A.; Dumas, E.; Mayer, C. R.; Mialane, P. Chem.Reν. 2010, 110, 6009. doi: 10.1021/cr1000578
(25)Izarova, N. V.; Pope, M. T.; Kortz, U. Angew. Chem., Int. Εd.2012, 51, 2. doi: 10.1038/srep25154
(26)Dablemont, C.; Hamaker, C. G.; Thouvenot, R.; Sojka, Z.;Che, M.; Maatta, E. A.; Proust, A. Chem. -Εur J. 2006, 12,9150. doi: 10.1002/chem.200600934
(27)Besson, C.; Musaev, D. G.; Lahootun, V.; Cao, R.;Chamoreau, L. M.; Villanneau, R.; Villain, F.; Thouvenot, R.;Geletii, Y. V.; Hill, C. L.; Proust, A. Chem. Εur. J. 2009, 15,10233. doi: 10.1002/chem.200900965
(28)Lahootun, V.; Besson, C.; Villanneau, R.; Villain, F.;Chamoreau, L. M.; Boubekeur, K.; Blanchard, S.; Thouvenot,R.; Proust, A. J. Am. Chem. Soc. 2007, 129, 7127.doi: 0.1021/ja071137t
(29)Sokolov, M. N.; Adonin, S. A.; Mainichev, D. A.; Sinkevich,P. L.; Vicent, C.; Kompankov, N. B.; Gushchin, A. L.;Nadolinny, V. A.; Fedin, V. P. Inorg. Chem. 2013, 52, 9675.doi: 10.1021/ic401492q
(30)Liu, C. G.; Liu, S.; Zheng, T. Inorg. Chem. 2015, 54, 7929.doi: 10.1021/acs.inorgchem.5b01002
(31)Coperet, C.; Comas-Vives, A.; Conley, M. P.; Estes, D. P.;Fedorov, A.; Mougel, V.; Nagae, H.; Nunez-Zarur, F.;Zhizhko, P. A. Chem. Reν. 2016, 116, 323.doi: 10.1002/chin.201615211
(32)Zhang, B.; Asakura, H.; Zhang, J.; Zhang, J.; De, S.; Yan, N.Angew. Chem. Int. Εd. 2016, 55, 8319-8323.doi: 10.1002/anie.201602802
(33)Zhang, B.; Asakura, H.; Yan, N. Ind. Εng. Chem. Res. 2017,56, 3578. doi: 10.1021/acs.iecr.7b00376
(34)Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101.doi: 10.1063/1.2370993
(35)Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 270.doi: 10.1063/1.448799
(36)Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299.doi: 10.1063/1.448975
(37)Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Reν. 2005, 105,2999. doi: 10.1021/cr9904009
(38)Glendening, A. E.; Carpenter, J. E.; Weinhold, F. ΝBO Version 3.1.
(39)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
(40)Zhang, B.; Asakura, H.; Zhang, J.; Zhang, I.; De, S.; Yan, N.Angew. Chem. Int. Εd. 2016, 55, 1.doi: 10.1002/anie.201602801
猜你喜歡
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
中國(guó)外匯(2019年17期)2019-11-16 09:31:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國(guó)資源綜合利用(2016年4期)2016-01-22 08:27:23
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
應(yīng)用化工(2014年3期)2014-08-16 13:23:50

- 物理化學(xué)學(xué)報(bào)的其它文章
- 基于概念密度泛函理論磷酸酯類(lèi)反應(yīng)性物質(zhì)毒性預(yù)測(cè)
- 從能量和信息理論視角理解單取代烷烴的異構(gòu)化
- 類(lèi)單晶硅結(jié)構(gòu)Si(C≡C―C6H4―C≡C)4新材料的力學(xué)與光學(xué)性質(zhì):第一性原理研究
- 嵌入配位不飽和金屬位對(duì)多孔芳香骨架材料儲(chǔ)氫性能的影響
- Strength of lntramolecular Hydrogen Bonds
- Fukui Functions for the Temporary Anion Resonance States of Be-, Mg-,and Ca-