硝基苯催化加氫合成對氨基苯酚的研究進展
2018-04-02 02:50:21劉迎新劉曉爽舒慧敏樓炯濤魏作君
石油化工 2018年1期
劉迎新,劉曉爽,曾 茂,舒慧敏,樓炯濤,魏作君
(1.浙江工業大學 藥學院 催化加氫研發基地,浙江 杭州 310014;2.浙江大學
生物質化工教育部重點實驗室 化學工程與生物工程學院 制藥工程研究所,浙江 杭州 310027)
對氨基苯酚(PAP)是一種重要的化工原料及有機中間體,廣泛應用于醫藥[1]、染料[2]、橡膠[3]等領域。PAP的合成工藝按原料路線可主要分為對硝基苯酚法[4-5]、對硝基氯化苯法[6]、苯酚法[7]、硝基苯(NB)法[8-9]等。其中以對硝基苯酚為原料的鐵粉還原法[4]是生產PAP最早的工藝路線,但該法成本高、污染嚴重,現已很少使用。以苯酚和對硝基氯苯為原料的制備工藝流程長,總收率低。NB電解還原法[9]是相對環保的工藝路線,但工藝設備投資大,且需耗費大量電力。而NB和對硝基苯酚的催化加氫合成工藝都具有流程較短、污染較少、能源消耗低等優點,但NB的價格更為低廉,更具有工業開發價值。以NB為原料催化加氫制備PAP的反應需要加氫催化劑和酸催化劑。加氫催化劑以貴金屬[10-13]為主,其中Pt基負載型催化劑最為常用,而Ni基催化劑[14-15]是貴金屬催化劑的良好替代品,關于雙金屬催化劑[16-18]的研究也在發展當中。近年來,為了減少反應造成的污染,研究者們逐漸用固體酸[19-23]、酸性離子液體[24-26]、CO2/H2O 酸性反應體系[26-27]來代替傳統的硫酸作為酸催化劑。此外,以固體酸負載金屬制備雙功能催化劑[28-30]也是近年來較有前景的研究方向。
本文從反應機理開始,對NB催化加氫制PAP中使用的加氫催化劑和酸催化劑以及雙功能催化劑進行比較分析,并對其應用前景進行展望。制備出具有更高催化加氫活性和酸催化活性的復合催化體系是今后研究的重點。
1 反應機理
NB在酸性環境中加氫制PAP的過程包含兩步反應。首先,吸附于催化劑表面的NB加氫生成中間體苯基羥胺(PHA);然后,PHA在酸性條件下進行Bamberger重排反應生成PAP。PHA若進一步深度加氫則被還原為苯胺(AN),其反應歷程如圖1所示[31]。詳細的Bamberger重排反應機理如圖2 所示[32]。
NB催化加氫制PAP涉及復雜的多相反應體系,包括氣相(H2)、固相(催化劑)、有機相(NB)和水相4相,如圖3所示[33]。首先NB被吸附在催化劑表面形成液滴,H2通過水/有機相界面進入液滴被吸附到催化劑表面,NB催化加氫生成中間體PHA。若PHA脫離催化劑遷移到酸性水相中,則重排生成PAP。若PHA停留在催化劑表面則會進一步加氫生成副產物AN。所以PAP的選擇性與催化劑對PHA的吸附作用有關,PHA越容易從催化劑表面解吸,越有利于重排反應進行[34]。
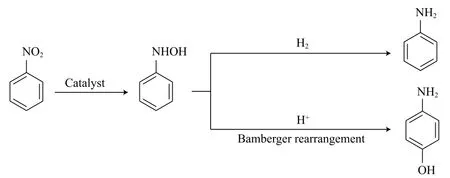
圖1 NB加氫制PAP的示意機理[31]Fig.1 Schematic mechanism for the hydrogenation of nitrobenzene(NB) to p-aminophenol(PAP)[31].
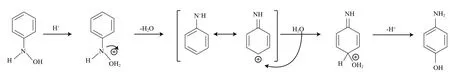
圖2 Bamberger重排機理[32]Fig.2 Bamberger rearrangement mechanism[32].
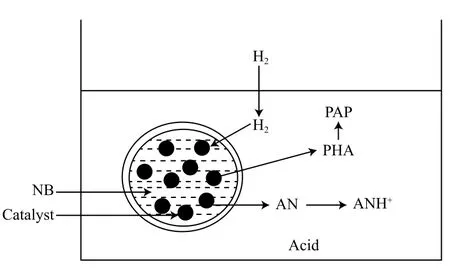
圖3 NB催化加氫的四相系統示意圖[33]Fig.3 Schematic representation of the four-phase system for the catalytic hydrogenation of NB[33].
2 加氫催化劑
在NB催化加氫合成PAP的反應中被用作加氫催化劑的活性組分多為貴金屬。通常情況下,Pt,Pd,Ru的負載型催化劑用于催化NB制備PAP的催化活性按 Pt > Pd > Ru 的順序依次遞減[34]。由于金屬Pt的吸附能力適中,使中間體PHA能順利進入酸性介質進行重排反應生成PAP,因而表現出較高的活性及選擇性。其他可催化NB加氫合成PAP 的貴金屬還有 Au[11]和 Ir[26]等。
由于貴金屬的成本較高,選用適當的載體來提高其催化活性和利用率是很有必要的。近年來NB催化加氫合成PAP的研究中較多采用Al2O3、活性炭和SiO2作為載體,其催化活性遞減順序為:活 性炭 > SiO2> Al2O3[35]。Al2O3作為 酸 堿 兩 性氧化物,具備一定的表面酸性,有利于提高PAP的選擇性;但Al2O3又具備一定的表面堿性,使表面酸性不至于過強,因而在CO2/H2O酸性反應體系中常使用Al2O3作為載體[17]。相對而言,活性炭和SiO2作為載體的適用范圍更廣。研究發現,負載在一些新型介孔碳材料CMK-1和CMK-3上的Pt催化劑的催化性能明顯好于Pt/C,說明新型介孔碳作為載體材料具有一定的優勢[12]。
2.1 單金屬催化劑
催化劑表面金屬粒徑的大小與PAP的選擇性之間存在緊密聯系。王淑芳等[36]制備了具有不同Pt粒徑的Pt/SiO2催化劑用于NB加氫合成PAP的反應。隨Pt粒徑的減小,NB轉化率及PAP選擇性均明顯提高,當Pt平均粒徑為5.1 nm時,NB轉化率達到100%,PAP選擇性達到48.5%。他們推測,隨Pt粒徑減小,催化劑表面的Pt原子暴露率增大,金屬活性位數量增加,造成NB轉化率增加。金屬活性位對中間產物PHA的吸附能力隨Pt粒徑減小而下降,導致PHA深度加氫生成AN的反應速率減慢,從而有效改善了重排反應與深度加氫副反應之間的競爭關系,提高了PAP選擇性。
金屬負載量的大小也與PAP的選擇性存在緊密聯系。研究表明,較低負載量的Pt催化劑具有較高的催化活性和選擇性。活性組分含量增大時,吸氫速率增大,繼而加快了中間體PHA深度加氫生成副產物AN的速度,PAP選擇性因此降低。Min等[12]發現,隨著Pt/CMK-1上Pt負載量從1%(w)升高到5%(w),NB的轉化率增加,但PAP的選擇性卻隨之降低。Nadgeri等[13]采用負載量1%(w)~5%(w)的Pt/C催化劑,在硫酸介質中催化NB制備PAP時,也發現隨著Pt負載量從1%(w)升高到3%(w),副產物AN的選擇性逐漸下降。而當Pt負載量升高到5%(w)時,AN的選擇性又升高,導致目標產物PAP的選擇性降低。
Ni基催化劑是用于催化NB制備PAP過程中貴金屬催化劑的良好代替品。Fu等[15]用氮化碳(CN)包覆Ni/Al2O3制備了CN/Ni/Al2O3催化劑,用于在硫酸中催化NB制PAP,轉化率可達80%,PAP選擇性可達100%。該課題組還發現,摻氮多孔碳包覆的鎳(Ni/CN)在硫酸溶液中催化NB加氫制備PAP也具有優良的催化性能和穩定性[14]。可能鎳為氮化碳和摻氮多孔碳提供了電子,使得它們具備吸附和活化氫的能力。同時Ni也因為被包覆而與反應環境隔離,避免了在強酸溶液中遭到腐蝕。該課題組還采用摻氮碳包覆的碳化鉬(CNMC-2)作為催化劑催化NB制備PAP,也表現出優良的活性和選擇性[37]。
金屬納米顆粒催化劑被用于NB催化加氫合成PAP的反應,表現出很好的活性。Dong等[38]制備了負載型鎳硅化物納米顆粒(Ni-Si/AC6)作為NB在硫酸中加氫制備PAP的催化劑,發現該催化劑具有優異的耐酸性和高活性,在多次重復使用后催化劑結構與催化活性仍保持穩定,且具有高PAP選擇性。該課題組用HZSM-5分子篩包覆Pt納米顆粒制成的雙功能催化劑也表現出高選擇性和穩定性[22]。
2.2 雙金屬催化劑
雙金屬催化劑主要通過金屬組分間的協同作用表現出與原金屬不同的電子和化學性能,在催化反應中具備更高的活性、選擇性和穩定性[39]。Zhang等[16]采用各種負載型Pt-Pb雙金屬催化劑(載體為活性炭、γ-Al2O3和SiO2),在加壓的CO2/H2O體系中催化NB制備PAP,并考察其催化性能。發現這些負載型Pt-Pb雙金屬催化劑比相同載體負載的Pt單金屬催化劑對PAP的選擇性要高,其中Pt-Pb/SiO2的催化性能最好。他們還采用Pt-Sn/Al2O3在加壓的CO2/H2O體系中催化NB制備PAP,發現在最佳條件下對PAP的選擇性高達85%[17]。Wang 等[18]將 Pt-Pb/MgAPO-5 催化劑用于NB催化加氫合成PAP,考察Pb的摻入對催化性能的影響。發現Pt-Pb/MgAPO-5催化劑上對PAP的選擇性要高于Pt/MgAPO-5。在最佳反應條件下NB的轉化率可達100%,PAP選擇性可達75%。在Pt/MgAPO-5催化劑中摻入Pb同時抑制了NB和PHA的氫化反應,導致PAP的選擇性有較高的提升。他們認為這可能是由于Pb和Pt的相互作用導致Pt表面的電子密度升高,有利于中間體PHA的解吸附。
3 酸催化劑
根據Bamberger重排機理,中間體PHA需在酸性催化劑作用下重排生成PAP,早期研究多以濃度較高的硫酸作為PHA重排的酸催化劑。研究表明,硫酸的最佳酸濃度為15%(w)左右,在該酸濃度下,以3%(w)Pt/C為加氫催化劑在最佳反應條件下催化NB加氫時,轉化率可達100%,PAP的選擇性可達75%[40]。雖然以硫酸等液體酸為酸性介質時催化活性較好,但過程中存在腐蝕設備、污染環境、后處理復雜、產品分離提純困難等諸多缺點。因此,后續研究者們采用固體酸、酸性離子液體、CO2/H2O酸性反應體系等來取代傳統液體酸,以減少過程對環境造成的污染。
3.1 固體酸
用固體酸代替傳統液體酸可降低廢液排放,且固體酸易于分離、無腐蝕作用、可重復使用、輸送安全、使用方便。硫酸化金屬氧化物作為固體超強酸是一類能夠實現強酸催化作用的環境友好型催化劑。Ratnam等[19]研究了一系列固體酸催化重排PHA制備PAP的催化性能。發現硫酸鋯金屬氧化物的催化性能最好,PAP的選擇性可高于90%。他們還將Pt基負載型催化劑和在500~650 ℃高溫下煅燒的Zr(SO4)2共混后用于催化NB制PAP。2%(w)Pt/ZrO2與Zr(SO4)2共混后作為催化劑時,在最佳條件下反應,NB轉化率可達97%,PAP的選擇性可達86%[20]。Wang等[28]發現,煅燒溫度對磺化的ZrO2固體酸的酸性和催化活性有較大影響。隨著煅燒溫度的升高,表面Br?nsted酸性位點數量減少、而Lewis酸性位點數量增加,高的煅燒溫度雖然降低了酸中心數目,但提高了其酸強度。Liu等[21]制備了復合金屬氧化物固體酸再結合Pt/ZrO2用于催化NB加氫重排制備PAP。他們發現隨著Al2O3加入量的增加,PAP的選擇性不斷增高。當Zr和Al的摩爾比為1∶9時,PAP選擇性可達74.9%,NB轉化率為44.8%。他們認為這可能是因為添加Al2O3后,Al、Zr和硫酸化劑相互作用形成新的Al—O—Zr鍵,使酸強度增高。隨著Al加入量的不斷增加,中等強度的酸性位點數量減少、且酸強度減弱,但出現新的表面強酸性位點。
硅鋁酸鹽分子篩具有孔道大小均勻、形態選擇性獨特、熱穩定性和水熱穩定性好、且強酸位可調等優點[41],也被作為酸催化劑用于NB加氫合成PAP。Wang等[23]分別在無氟體系和含氟體系中合成了一系列磷酸硅鋁分子篩(SAPO-5),將其與Pt/SiO2混合作為NB加氫合成PAP的催化劑。發現由于表面強酸性位點的出現,含氟體系制備的SAPO-5分子篩的催化性能明顯比無氟體系制備的分子篩更好,具有更高的PAP產率。他們認為,HF的存在促進了Si在分子篩結構中的插入和Si配體環境的形成,導致分子篩酸量降低,但酸強度增加,分子篩表面酸強度的增加促進了重排反應的酸催化性能。
阻礙固體酸進一步應用的主要原因是其穩定性較差。磺酸化的碳基固體酸具有很高的表面酸位密度和熱穩定性,且其表面具有良好的疏水性,酸活性位點不會被水溶解而流失,導致化學穩定性較好;并且碳基固體酸價格低廉,成本較低,因而受到了越來越多的關注[42]。本課題組[8]以廉價的碳水化合物為碳源,采用一步水熱碳化法制備了一系列碳基固體酸,以Pt/C為加氫催化劑,用于液相催化NB制備PAP。研究發現,在碳基材料磺化后所引入的強酸性—SO3H基團是主要的酸催化活性位。其中以淀粉為碳源制備的碳基固體酸表面酸性最強,作為酸催化劑時生成PAP的選擇性最高。該復合催化體系在最佳反應條件下,PAP選擇性為77.8%,NB轉化率為61%。白玉婷等[43]將Ni/SiO2與磺酸功能化的C/SBA-15固體酸(SO3H-C/SBA-15)混合用于催化NB制備PAP,并系統考察了催化劑組成對催化性能的影響。他們發現Ni/SiO2上的活性中心分布均勻,碳含量為40%的SO3H-C/SBA-15催化劑上的表面酸值最大。當Ni/SiO2和SO3H-C/SBA-15質量比為1∶6時,催化NB制備PAP的活性較好。
3.2 酸性離子液體
功能化Br?nsted酸性離子液體兼具液體酸流動性好和酸強度分布均勻,以及固體酸無揮發和低腐蝕的特點。在酸催化反應中具有活性高、易于分離、可循環使用及環境友好等優點,表現出很大的應用潛力[44]。崔詠梅等[24]采用 Pt/SiO2和季銨型Br?nsted 酸性離子液體[HSO3-b-N(CH3)3]HSO4構成的催化體系催化NB制備PAP。在最佳條件下反應,NB轉化率為96.6%,PAP的選擇性為81.4%。[HSO3-b-N(CH3)3]HSO4因含 有2 個質子位和較高的酸強度,對PHA重排制備PAP的反應具有良好的催化性能。由于使用物理方法固載的離子液體易流失,可采用化學鍵合的方法將離子液體固載于硅膠表面。該課題組將酸性離子液體用化學鍵固定于硅膠表面,制備了[HSO3-bvim]HSO4/SiO2固體酸催化劑。但由于硅膠表面存在的羥基數目有限,再加上空間位阻的作用,造成表面接枝的酸性離子液體的數量很少,不足以為Bamberger重排反應提供足夠的酸催化中心數量[25]。
Wang等[26]將 Ir/C和一系列功能化Br?nsted 酸性離子液體組成的催化體系應用于NB加氫制PAP的研究中。其中[HSO3-bmim]HSO4在加入表面活性劑的情況下結合Ir/C用于催化NB加氫反應,轉化率可達100%,PAP的選擇性為62.4%。雙功能化離子液體[HSO3-b-N-Bu3]HSO4由于具有較高的親脂性,在用作酸催化劑的同時也可用作表面活性劑。這使得該催化體系具有獨特的優勢。
3.3 CO2/H2O酸性體系
CO2是一種無毒、安全無害、來源廣泛、價格低廉的常用氣體。CO2在H2O中發生電離平衡時,可形成原位碳酸,過程機理如圖4所示。該體系在一定溫度和壓力下的pH可達3,目前已成為一種備受關注的傳統無機酸替代品[45]。Weikel等[46]研究了CO2/H2O體系中形成的原位酸催化劑,在三種溶劑系統中提供原位酸進行催化。在反應結束后,體系可以很容易地通過降壓除去CO2,不需要中和。從而避免使用堿來中和酸的過程,減少了含鹽廢水的排放,以達到環境友好。
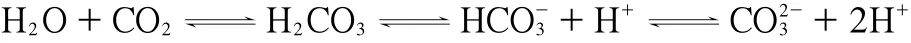
圖4 CO2 在水中發生電離平衡的重排機理[45]Fig.4 The rearrangement mechanism of CO2 ionization equilibrium in water[45].
Liu等[27]研究了 PHA 在 CO2/H2O 體系中的Bamberger重排反應,在最佳條件下反應1 h,PAP的產率達到80%。之后該課題組采用Pt-Sn/Al2O3催化劑在加壓的CO2/H2O體系中催化NB加氫制備PAP,PAP的選擇性可高達85%[17]。他們還采用多種負載型Pt-Pb雙金屬催化劑,在加壓的CO2/H2O體系中催化NB加氫制備PAP,Pt-Pb/SiO2的催化性能最好[16]。Zhao等[35]在超臨界CO2/H2O體系中,使用Pt/Al2O3催化劑催化NB加氫制備PAP。他們發現隨著CO2壓力的增加,NB的轉化率降低,而PAP的選擇性增高。可能因為CO2與H2反應產生的CO占據了催化劑表面Pt的低配位點,從而抑制了NB的加氫和PHA的深度加氫。該體系中NB轉化率可高達98.5%,PAP產率可達68.9%。
4 雙功能催化劑
通過將加氫活性組分與表面酸性位點結合制備雙功能催化劑(如將金屬Pt負載在固體酸上),使得NB催化加氫合成PAP的研究有了進一步的發展。Wang等[28]制備了金屬-固體酸雙功能催化劑Pt-S2O82-/ZrO2催化NB加氫制備PAP,得到的PAP最高產率為23.9%。他們通過減少Pt的負載量來抑制催化加氫活性,可以提高在酸性位點上PHA重排反應的競爭性,從而使PAP選擇性更高。他們還制備了Pt-[HSO3-bvim]HSO4/SiO2雙功能催化劑,該催化劑對NB加氫制PAP的反應具有一定的催化活性,但PAP產率過低[25]。該課題組還用Mg改性的磷酸鋁分子篩(MgAPO-5)作為載體,制備了Pt/MgAPO-5雙功能催化劑并考察其催化活性[29]。結果表明,MgO/Al2O3的比率影響Mg的含量和MgAPO-5試樣的酸性,因此在很大程度上影響了Pt/MgAPO-5的催化活性。而Pt/MgAPO-5對PAP的選擇性由MgAPO-5中強酸性位點的數量所決定。當MgO/Al2O3的摩爾比為0.5時,MgAPO-5試樣顯示出最大數量的表面強酸性位點,并且在Pt/MgAPO-5上達到最高的PAP產率(41.1%)。
本課題組制備了一系列Pt-碳基固體酸雙功能催化劑,并考察其對NB加氫制PAP反應的催化性能[30]。其中,以淀粉為碳源制備的碳基固體酸表面酸性最強,在用其負載Pt所制備的雙功能催化劑作用下,NB轉化率最高可達93.6%,但PAP的選擇性較低為23.4%,催化性能尚不夠理想。還制備了磺化石墨烯固體酸催化劑,發現它具有與硫酸相比擬的酸催化活性[47]。氧化石墨烯被廣泛用作加氫催化劑載體,且氧化石墨烯負載金屬催化劑[48]往往具有更好的催化活性和選擇性。因此,將金屬負載在石墨烯固體酸上制備新型雙功能催化劑,用于催化NB加氫制PAP或將成為另外一個新的研究方向。
將用固體酸包覆的金屬納米顆粒作為NB催化加氫制PAP的雙功能催化劑是另一個較新的研究思路。Gu等[22]用HZSM-5硅鋁酸鹽分子篩包覆Pt納米顆粒制成雙功能催化劑(Pt@HZSM-5),將其應用于NB催化加氫合成PAP的反應中,發現該催化劑體系具有良好的催化性能,NB產率可達100%,PAP選擇性可達75%,而且催化劑有良好的可回收性。該雙功能催化劑有別于其他催化劑的地方在于,Pt納米顆粒與酸性位點之間存在協同作用。金屬Pt位點被酸性位點緊密包圍時可促進PHA重排生成PAP,而與Pt納米顆粒距離較遠的表面酸性位點對PHA重排反應基本是無效的。
5 結語
現階段NB催化加氫合成PAP的研究主要有兩條思路:一是以金屬為加氫催化劑,與固體酸、酸性離子液體、CO2/H2O酸性反應體系等構成催化體系;二是制備加氫組分與酸性位點結合的雙功能催化劑。今后可尋找比硅膠更合適的材料用于固定酸性離子液體,并進一步負載金屬得到雙功能催化劑;也可進一步制備不同類型的雙金屬負載型催化劑與酸催化劑構成復合催化體系,或將雙金屬組分負載于固體酸材料上制備雙功能催化劑。嘗試用硅鋁酸鹽分子篩包覆不同類型的金屬納米顆粒來制備雙功能催化劑也是一個值得深入研究的方向。
[1] Bertolini A,Ferrari A,Ottani A,et al. Paracetamol:New vistas of an old drug[J].CNS Drug Rev,2006,12(3/4):250-275.
[2] Lerner L. Identity of a purple dye formed by peroxidic oxidation ofp-aminophenol at low pH[J].J Phys Chem A,2011,115(35):9901-9910.
[3] El-Wakil A A. Amelioration of acrylonitrile-butadiene copolymer properties using natural rubber graftp-aminophenol[J].J Appl Polym Sci,2010,104(1):27-34.
[4] Lai Bo,Chen Zhaoyu,Zhou Yuexi,et al. Removal of high concentrationp-nitrophenol in aqueous solution by zero valent iron with ultrasonic irradiation(US-ZVI)[J].J Hazard Mater,2013,250/251:220-228.
[5] Vaidya M J,And S M K,Chaudhari R V. Synthesis ofp-aminophenol by catalytic hydrogenation ofp-nitrophenol[J].Org Process Res Dev,2002,7(2):202-208.
[6] 楊穎,菅盤銘. 對氨基苯酚連續水解催化加氫合成工藝的研究[J].廣東化工,2012,39(12):89-90.
[7] 劉竹青,胡愛琳,王公應. 對氨基苯酚的合成研究進展[J].工業催化,1999,7(2):11-16.
[8] Liu Yingxin,Fang Yanyan,Lu Xiaolei,et al. Hydrogenation of nitrobenzene top-aminophenol using Pt/C catalyst and carbon-based solid acid[J].Chem Eng J,2013,229(8):105-110.
[9] Polat K,Aksu M L,Pekel A T. Electroreduction of nitrobenzene top-aminophenol using voltammetric and semipilot scale preparative electrolysis techniques[J].J Appl Electrochem,2002,32(2):217-223.
[10] Quartarone G,Ronchin L,Tosetto A,et al. New insight on the mechanism of the catalytic hydrogenation of nitrobenzene to 4-aminophenol in CH3CN-H2O-CF3COOH as a reusable solvent system. Hydrogenation of nitrobenzene catalyzed by precious metals supported on carbon[J].Appl Catal,A,2014,475(5):169-178.
[11] Zou Luyao,Cui Yuanyuan,Dai Weilin. Highly ef fi cient Au/TiO2catalyst for one-pot conversion of nitrobenzene top-aminophenol in water media[J].Chin J Chem,2014,32(3):257-262.
[12] Min K I,Choi J S,Chung Y M,et al.p-Aminophenol synthesis in an organic/aqueous system using Pt supported on mesoporous carbons[J].Appl Catal,A,2008,337(1):97-104.
[13] Nadgeri J M,Biradar N S,Patil P B,et al. Control of competing hydrogenation of phenylhydroxylamine to aniline in a single-step hydrogenation of nitrobenzene top-aminophenol[J].Ind Eng Chem Res,2011,50(9):5478-5484.
[14] Wang Tao,Dong Zhen,Fu Teng,et al. Nickel embedded in N-doped porous carbon for the hydrogenation of nitrobenzene top-aminophenol in sulphuric acid[J].Chem Commun,2015,51(100):17712-17715.
[15] Fu Teng,Wang Meng,Cai Weimeng,et al. Acid-resistant catalysis without use of noble metals:Carbon nitride with underlying nickel[J].ACS Catal,2014,4(8):2536-2543.
[16] Zhang Tingting,Jiang Jingyang,Wang Yanhua. Supported bimetallic catalyst Pt-Pb/SiO2for selective conversion of nitrobenzene top-aminophenol in pressurized CO2/H2O system[J].Chin Chem Lett,2017,28(2):307-311.
[17] Zhang Tingting,Jiang Jingyang,Wang Yanhua. Green route for the preparation ofp-aminophenol from nitrobenzene by catalytic hydrogenation in pressurized CO2/H2O system[J].Org Process Res Dev,2015,19(12):2050-2054.
[18] Wang Shufang,He Beibei,Wang Yanji,et al. MgAPO-5-supported Pt-Pb-based novel catalyst for the hydrogenation of nitrobenzene top-aminophenol[J].Catal Commun,2012,24:109-113.
[19] Ratnam K J,Reddy R S,Sekhar N S,et al. Bamberger rearrangement on solid acids[J].Appl Catal,A,2008,348(1):26-29.
[20] Deshpande A,Figueras F,Kantam M L,et al. Environmentally friendly hydrogenation of nitrobenzene top-aminophenol using heterogeneous catalysts[J].J Catal,2010,275(2):250-256.
[21] Liu Pingle,Hu Yaohua,Ni Min,et al. Liquid phase hydrogenation of nitrobenzene topara-aminophenol over Pt/ZrO2catalyst and SO42-/ZrO2-Al2O3solid acid[J].Catal Lett,2010,140(1):65-68.
[22] Gu Jing,Zhang Zhiyang,Ding Liping,et al. Platinum nanoparticles encapsulated in HZSM-5 crystals as an ef fi cient catalyst for green production ofp-aminophenol[J].Catal Commun,2017,97:98-101.
[23] Wang Shufang,Wang Yanji,Yang Gao,et al. Preparation of SAPO-5 and its catalytic synthesis ofp-aminophenol[J].Chin J Catal,2010,31(6):637-644.
[24] 崔詠梅,袁達,王延吉,等. 酸性離子液體作催化劑的硝基苯加氫合成對氨基苯酚[J].化工學報,2009,60(2):345-350.
[25] 崔詠梅,王淑芳,趙新強,等. Pt-[HSO3-bvim]HSO4/SiO2雙功能催化劑制備及其催化性能研究[J].高校化學工程學報,2009,23(4):617-622.
[26] Wang Hong,Jiang Taotao,Ma Lei,et al. Ir/C and Br?nsted acid functionalized ionic liquids:An ef fi cient catalytic system for hydrogenation of nitrobenzene top-aminophenol[J].RSC Adv,2017,50 (7):31663-31670.
[27] Liu Shijuan,Hao Yuanping,Jiang Jingyang. Bamberger rearrangement ofN-arylhydroxylamine top-aminophenol in a CO2-H2O system[J].Ind Eng Chem Res,2014,53(20):8372-8375.
[28] Wang Shufang,Ma Yuanhui,Wang Yanji,et al. Synthesis ofp-aminophenol from the hydrogenation of nitrobenzene over metal-solid acid bifunctional catalyst[J].J Chem Technol Biotechnol,2008,83(11):1466-1471.
[29] Wang Shufang,Jin Yadan,He Beibei,et al. Synthesis of bifunctional Pt/MgAPO-5 catalysts and their catalytic performance in the hydrogenation of nitrobenzene top-aminophenol[J].Sci Chin Chem,2010,53(7):1514-1519.
[30] 劉迎新,方艷艷,李喜英,等. 碳基固體酸負載鉑雙功能催化劑的制備及其對硝基苯加氫制對氨基苯酚催化性能的研究[J].浙江工業大學學報,2013,41(1):48-52.
[31] Rode C V,Vaidya M J,Jaganathan R,et al. Hydrogenation of nitrobenzene top-aminophenol in a four-phase reactor:Reaction kinetics and mass transfer effects[J].Chem Eng Sci,2001,56(4):1299-1304.
[32] Kurc L,Cerveny L,Toman J. Manufacture of 4-aminophenol by catalytic hydrogenation of nitrobenzene[J].Chem Prum,1992,42(4):85-89.
[33] Tanielyan S K,Nair J J,Marin N,et al. Hydrogenation of nitrobenzene to 4-aminophenol over supported platinum catalysts[J].Org Process Res Dev,2007,11(4):681-688.
[34] Juang T M,Hwang J C,Ho H O,et al. Selectivity in the phase transfer catalytic hydrogenation of nitrobenzene by transition Ⅷ metals[J].J Chin Chem Soc,1988,35(2):135-140.
[35] Zhao Lijun,Cheng Haiyang,Liu Tong,et al. A green process for production ofp-aminophenol from nitrobenzene hydrogenation in CO2/H2O:The promoting effects of CO2and H2O[J].J CO2Util,2017,18:229-236.
[36] 王淑芳,高楊,王延吉,等. 負載型納米Pt催化劑的制備及其催化合成對氨基苯酚[J].石油化工,2009,38(4):361-366.
[37] Wang Tao,Dong Zhen,Cai Weimeng,et al. An ef fi cient hydrogenation catalyst in sulfuric acid for the conversion of nitrobenzene top-aminophenol:N-doped carbon with encapsulated molybdenum carbide[J].Chem Commun,2016,52(70):10672-10675.
[38] Dong Zhen,Wang Tao,Zhao Jie,et al. Ni-silicides nanoparticles as substitute for noble metals for hydrogenation of nitrobenzene top-aminophenol in sulfuric acid[J].Appl Catal,A,2016,520(25):151-156.
[39] Yu Weiting,Porosoff M D,Chen Jingguang. Review of Ptbased bimetallic catalysis:From model surfaces to supported catalysts[J].Chem Rev,2012,112(11):5780-5817.
[40] Rode C V,Vaidya M J,Chaudhari R V. Synthesis ofp-aminophenol by catalytic hydrogenation of nitrobenzene[J].Org Process Res Dev,1999,3(6):465-470.
[41] Cundy C S,Cox P A. The hydrothermal synthesis of zeolites:History and development from the earliest days to the present time[J].Chem Rev,2003,103(3):663-702.
[42] Hara M,Yoshida T,Takagaki A,et al. A carbon material as a strong protonic acid[J].Angew Chem,Int Ed,2004,43(22):2955-2958.
[43] 白玉婷,朱學成,張利雄,等. Ni/SiO2和SO3H-C/SBA-15復合催化劑上硝基苯加氫制對氨基苯酚[J].催化學報,2013,34(1):263-271.
[44] Zhu Huaping,Yang Fan,Tang Jie,et al. Br?nsted acidic ionic liquid 1-methylimidazolium tetrafluoroborate:A green catalyst and recyclable medium for esterification[J].Green Chem,2003,5(1):38-39.
[45] Roosen C,Ansorge-Schumacher M,Mang T,et al. Gaining pH-control in water/carbon dioxide biphasic systems[J].Green Chem,2007,9(5):455-458.
[46] Weikel R R,Hallett J P,Liotta C L,et al. Self-neutralizing in situ acid catalysts from CO2[J].Top Catal,2006,37(2):75-80.
[47] Thushara D,潘若飛,賀曉東,等. 磺化石墨烯固體酸催化劑的制備及活性評價[J].工業催化,2014,22(9):676-679.
[48] Wei Zuojun,Pan Ruofei,Hou Yaxin,et al. Graphenesupported Pd catalyst for highly selective hydrogenation of resorcinol to 1,3-cyclohexanedione through giantπ-conjugate interactions[J].Sci Rep,2015,5(23):15664-15672.
猜你喜歡
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
應用化工(2014年3期)2014-08-16 13:23:50
