Bonding and Reactivity in RB-AsR Systems (R = H, F, OH, CH3, CMe3,CF3, SiF3, BO): Substituent Effects
2018-03-29 03:12:17GHARAManasCHATTARAJPratim
物理化學學報 2018年2期
GHARA Manas, CHATTARAJ Pratim K.*
Department of Chemistry and Center for Theoretical Studies, Indian Institute of Technology, Kharagpur 721302, India.
1 Introduction
The incessant quest towards the synthesis of molecules containing multiple bonds involving main group elements has been an active field of research1. In this context, the triply bonded RE-ER (E = Group 14 element) systems are well synthesized and experimentally characterized by many research groups2–13. Since, the replacement of Group 14 elements by one Group 13 element and one Group 15 element is possible in RE-ER provided the substituents {R} are judiciously chosen. In this way triply bonded RB-NR systems were reported by Paetzold14. On the other hand, in 1990 Power and coworkers15have synthesized borylarsinide anion [PhAs-BMes2]?, which contains an As―B bond with the distance of 1.936 ? (1 ? =0.1 nm) suggesting an As=B double bond. A few years later, in 2006 the same group has prepared P=B and As=B double bonded compounds by making use of the donor stabilization strategy16. In 1993, Jones and coworkers17synthesized monomeric arsinoboranes, where the B―As bond order was estimated to be 1.6. However, in 1989 Nguyen and coworkers18and in 1996 Watts and coworkers19have done computational studies on HPBH system, where they have shown that the most stable isomer of the system has a bent geometry with a B―P―H bond angle of 94.5° and the B―P bond length of 1.756 ? having a B=P double bond character. Recently, the group of Su has shown the substituent effects on the stability of RB-SbR20, RB-BiR21and RIn-AsR22systems and they have also predicted the presence of triple bond between those Gr-13 and a Gr-15 element although it is very weak. In the present work we study the substituent effects on the stability and the nature of bonding in RB-AsR systems. In addition, the effect of substituents23on the chemical reactivity and selectivity of these systems are also characterized using global and local reactivity descriptors obtained from the conceptual density functional theory24,25.
2 Theoretical background
Conceptual density functional theory24,25based reactivity descriptors and the associated popular qualitative electronic structure principles provide qualitative trends in different systems and processes by unifying experimental/ calculated data even though they are empirical in nature. The electrophilicity (ω), a global reactivity index which measures the propensity of a species to accept electrons, as proposed by Parr et al.26is defined as27
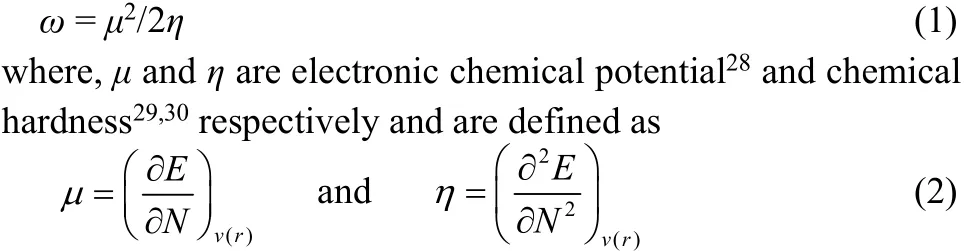
Applying a finite difference approximation, the above expressions can be written as
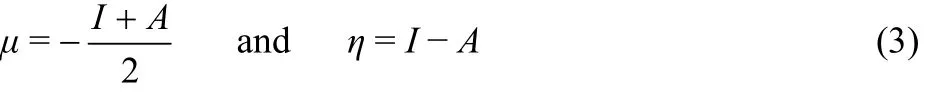
where I and A are the first vertical ionization potential and first vertical electron affinity respectively and these are determined by ΔSCF finite difference approach (FDA), where I and A for an N-electron system are expressed as

where E(N), E(N ? 1) and E(N + 1) are electronic energies of N,(N ? 1) and (N + 1) electronic systems respectively. A related qualitative electronic structure principle is electrophilicity equalization principle a formal analytical proof31of which was provided assuming the simultaneous validity of two hitherto well accepted principles, viz., electronegativity and hardness equalization principles32–34. Even the term ‘principle’35is used in standard text books for cases like the Le Chatelier principle even when there are known exceptions36.
On the other hand, the local reactivity descriptors such as the Fukui function as proposed by Parr and Yang37, can be written as

The discontinuous nature of ρ(r) vs N plot gives three types of Fukui functions,

where ρN(r), ρN?1(r) and ρN+1(r) are electron densities of N, (N ?1) and (N + 1) electronic systems respectively.
Yang and Mortier38proposed ‘condensed’ Fukui functions on each atomic site k in a molecule as

where qkis the electronic population of k-th site in a molecule.
Chattaraj et al.39,40proposed another global reactivity descriptor called philicity which includes almost all global and local information of a molecule as well as the electrophilic and nucleophilic power of an atomic site in a molecule, which is defined as

where, α = +, ? and 0 are for nucleophilic, electrophilic and radical attacks respectively.
In 2005 Toro-Labbe and co-workers41,42introduced a dual descriptor (Δf(r)), which is expressed as

In 2007 Chattaraj and co-workers43recommended another reactivity descriptor, called multiphilicity which identifies both the electrophilic and nucleophilic characteristics of an atom in a molecule. This is defined as

when Δωk> 0, the k-th site is favorable for nucleophilic attack and if Δωk< 0, the k-th site is favorable for electrophilic attack.Note that the Fukui function and the dual descriptor are intramolecular in nature whereas the multiphilicity is an intermolecular reactivity descriptor. A related global descriptor is net electrophilicity44.
3 Computational details
Optimization of geometries of all the studied molecules has been carried out at the DFT level of theory using the exchange-correlation energy functional M06-2X45. A triple-ζ quality basis set augmented by a single polarization function,def2-TZVP has been employed as atomic orbital basis function for these calculations. Harmonic vibrational frequency analysis has been performed in order to characterize the nature of stationary points on the potential energy surface and to determine zero point energy correction. All these calculations have been done using Gaussian 09 programme46suit. Natural bond orbital (NBO)47analysis is performed in order to get natural electronic population48on each atomic site which gives atomic charge and Wiberg bond index (WBI)49values for a chemical bond. This calculation is carried out using NBO 5.0 software package as embedded in Gaussian 09 software.Electron density analysis (EDA)50is also performed to understand the nature of bonding using Multiwfn softwere.51

Fig.1 Bonding scheme for the RB-AsR systems.
4 Results and discussion
Optimization reveals that the RB-AsR system has a bent geometry as shown in Fig.1. There are two possible bonding models in order to interpret the bonding situation in these species, which are pictorially depicted in Fig.1. Here, we have taken the R group as H, F, OH, CH3, CMe3, CF3, SiF3and BO.One may consider that the formation of RB-AsR takes place through the combination of two fragments RB and AsR. Our calculation shows that the ground states (G.S.) of RB and AsR are singlet and triplet respectively. We also calculate the energy of the triplet excited state (E.S.) of RB and that of the singlet E.S. of AsR. Thus, we get the singlet-triplet splitting energies(ΔEST) of RB and AsR fragments which are given in Table 1.Now, if the magnitude of ΔESTof RB is greater than that of AsR fragment then Scheme-I one is favorable and if the reverse is true then Scheme-II is favorable. The difference between Schemes-I and II is that there is a possibility of one B → As σ and two B ← As π bond in Scheme-I whereas Scheme-II comprises two σ and π bonds and one B ← As π bond.
The important geometrical parameters of RB-AsR systems are listed in Table 1 and the optimized geometries are given in Fig.2. The ∠B―As―R(°) bond angles demonstrate that all the systems exhibit a bent geometry as shown in Fig.1. Whereas,the ∠R―B―As(°) bond angles are nearly 180°. The experimental data available for B―As bond length is 1.936 ? in borylarsinide anion15[PhAs-BMeS2]?and 1.914 ? in boranylidenearsane Ar*As-B(DMAP)Tmp16. Thus, our calculated values of B―As bond length show that there should be double bond between these two atoms and even with slightly triple bond character in some cases as highlighted by the corresponding Wiberg bond index values as shown in the next section. The ΔEST values of RB and AsR fragments show that Scheme-I is favorable for FB-AsF, HOB-AsOH, H3CB-AsCH3and Me3CB-AsCMe3systems and Scheme-II is favorable for HB-AsH, F3CB-AsCF3, F3SiB-AsSiF3and OBB-AsBO systems. We have also calculated the values of HOMO-LUMO energy gap (H-L Gap) in these systems (see Table 1). We see that the H-L Gap is greater in those systems which follow Scheme-II and thus showing greater stability in comparison to those systems which follow Scheme-I. This was expected because there are two classical σ and π bonds and one B ← As π bond in Scheme-II where as in Scheme-I all the bonds are coordinate covalent type. Since the lone pairs in AsR consist of the valence s- and one of the valence p- orbitals of As effective overlap will not take place with an orbital of B atom having different size.
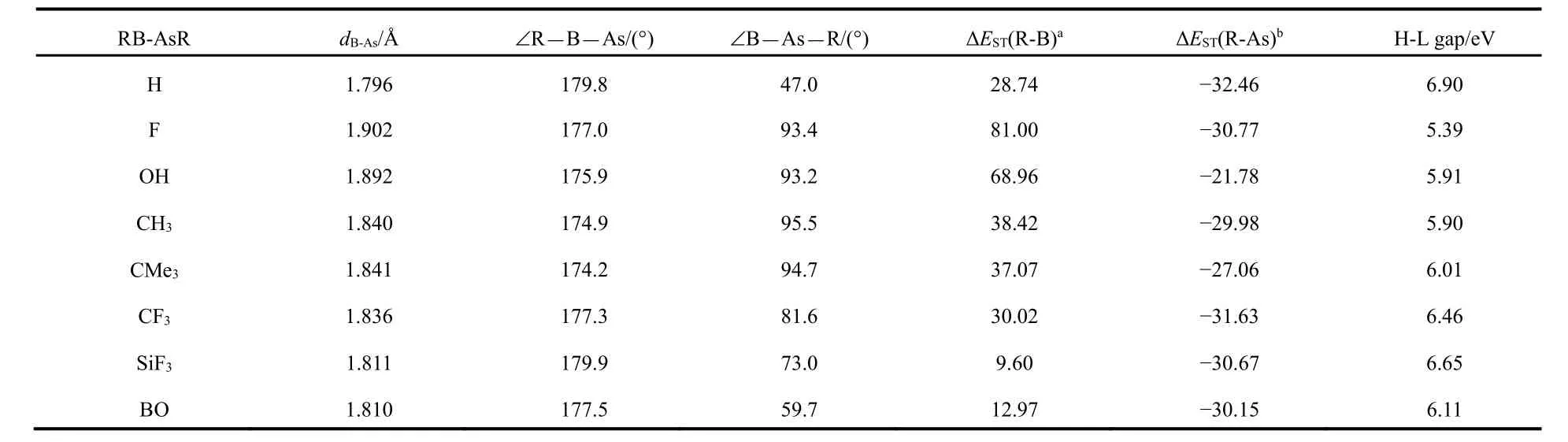
Table 1 The geometrical parameters, singlet-triplet splitting energy of R-B (ΔEST(R-B)) and R-As (ΔEST(R-As)) and HOMO-LUMO gap (eV) at M06-2X/def2-TZVP lavel.
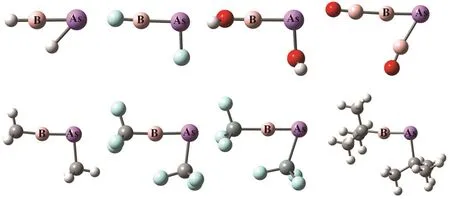
Fig.2 The optimized geometries of RB-AsR systems (R = H, F, OH, BO, CH, , CF, SiF and CMe) at the M06-2X/def2-TZVP level.
We have examined the stability of RB-AsR systems by comparing the two isomers R2B-As and B-AsR2 on the singlet potential energy surface which are given in Fig.3. The study reveals that RB-AsR systems belong to the global minima on the respective potential energy surfaces for F3SiB-AsSiF3,Me3CB-AsCMe3and HB-AsH systems. Note that the relative energy of the H2B-As is slightly greater (0.0044 kcal·mol?1, 1 cal = 4.1868 J) than that of the HB-AsH isomer. On the other hand R2B-As are the global minima in case of R = CH3, CF3,OH and F. Moreover, in case of RB-SbR systems (R = H, F,OH, CH3and SiH3) as reported by the group of M. D. Su20, all the R2B-Sb isomers are more stable in comparison to RB-SbR.Whereas, in case of HBPH system as reported by Nguyen18,H2B-P isomer is less stable in comparison to HB-PH one.However, for OBB-AsBO system we did not obtain the minimum energy structure on the potential energy surface which corresponds to R2B-As at M06-2Xdef2-TZVP level.But, we could get that at HF/3-21G level, which indicates that the barrier between (OB)2B-As and OBB-AsBO is very small at the higher level. A similar situation was also observed before for HB-PH system.18
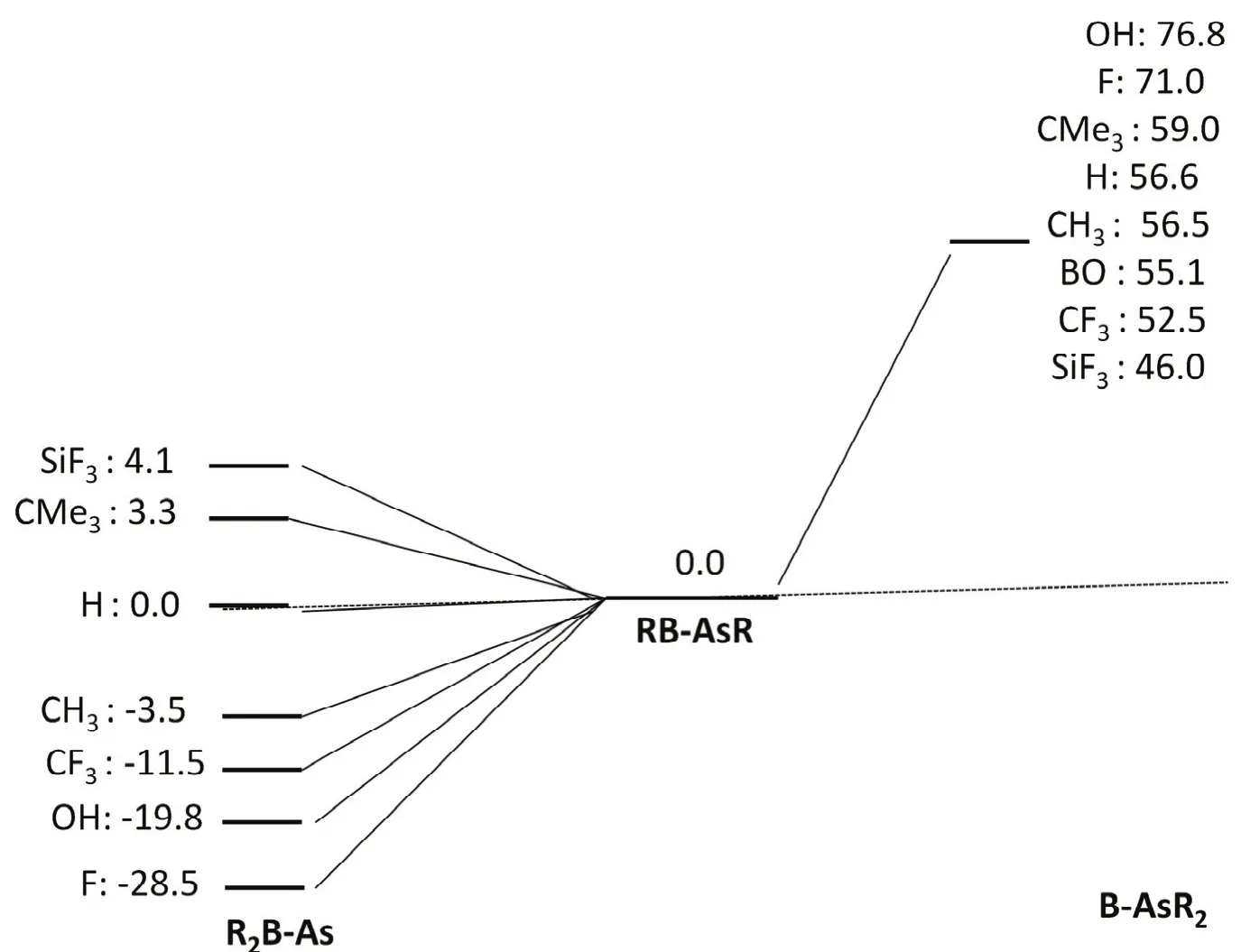
Fig.3 Relative energy values for RB-AsR (R = F, OH, CF3, CH3, H, CMe3 BO and SiF3) in kcal·mol?1 calculated at the M06-2X/def2-TZVP level.
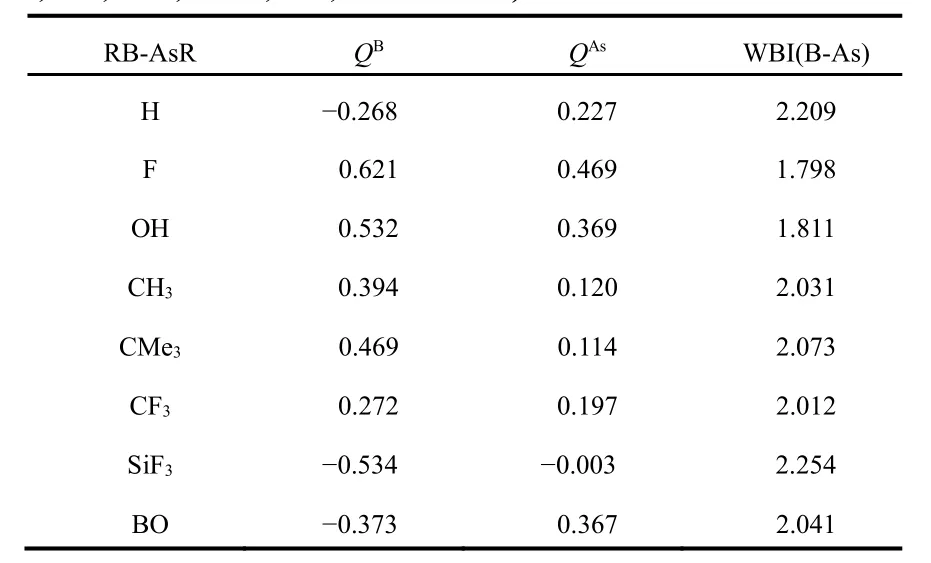
Table 2 The NPA charges on B and As centres (Q, a.u.) and Wiberg bond index (WBI) values of B-As bonds in RB-AsR (R=H,F, OH, CH3, CMe3, CF3, SiF3 and BO) at M06-2X/def2-TZVP level.
4.1 Bonding analysis
In order to understand the nature of bonding between B and As centers in these systems we have calculated NPA charges on B and As centers and WBI values in between them, which are given in Table 2. The NPA charges on B and As centers show that the bonding between B and As centers is not purely covalent and some ionic character is also there. The WBI values in case of FB-AsF (1.798) and HOB-AsOH (1.811) are less than two, whereas in all other cases it is greater than two.For comparison we calculated WBI of ethylene (2.049) and acetylene (2.999) in the same level of theory. Comparing these values we may conclude that some triple bond character is present between B and As centers in HB-AsH, Me3CB-AsCMe3and F3SiB-AsSiF3systems. Since WBI values are 2.209, 2.073 and 2.254 in HB-AsH, Me3CB-AsCMe3and F3SiB-AsSiF3respectively although these are weak in comparison to that of acetylene. Presumably the reason for the formation of this weak triple bond is due to “inert s-pair effect” and “orbital non-hybridization effect”52,53.
4.2 Electron density analysis
We have analyzed the nature of bonding between B and As centers in these systems using electron density analysis technique. Bader’s atoms-in-a-molecule50method is used to analyze various parameters at the bond critical point (BCP) in between B and As atoms to characterize the nature of the B―As bond. The values of the topological parameters obtained at the BCP of B―As bonds are provided in Table 3. Generally higher value of electron density (ρ(rc)) and negative value of Laplacian of electron density (▽2ρ(rc)) at the BCP satisfy the nature of a typical covalent bond. But this is not always true,e.g. in F2molecule the value of (▽2ρ(rc)) is positive although the F―F bond in F2is typically covalent. So, other parameters like local kinetic energy density (G(rc)), local potential energy density (V(rc)), local electron energy density (H(rc)) and the ratio ?G(rc)/V(rc) are also calculated at the BCP of the B―As bond. From the table we see that the values of both (▽2ρ(rc))and (H(rc)) are negative at the BCP for all B―As bonds indicating the covalent character. Thus, analyzing the electron density descriptors we may conclude that the interaction between B and As atoms in these systems are of purely covalent type. We have also obtained another BCP in between B center and the H atom bent towards the B center in HB-AsH(see Fig.2).This bond is also of covalent type as confirmed by the electron density analysis. However, we did not get any BCP in between As and H centers.
The plots of (▽2ρ(r)) are provided in Fig.4. The color code is red: (▽2ρ(r)) < 0 and green: (▽2ρ(r)) > 0. The accumulation of electron density is depicted by the region with negative(▽2ρ(r)) values. It highlights the associated bond paths.
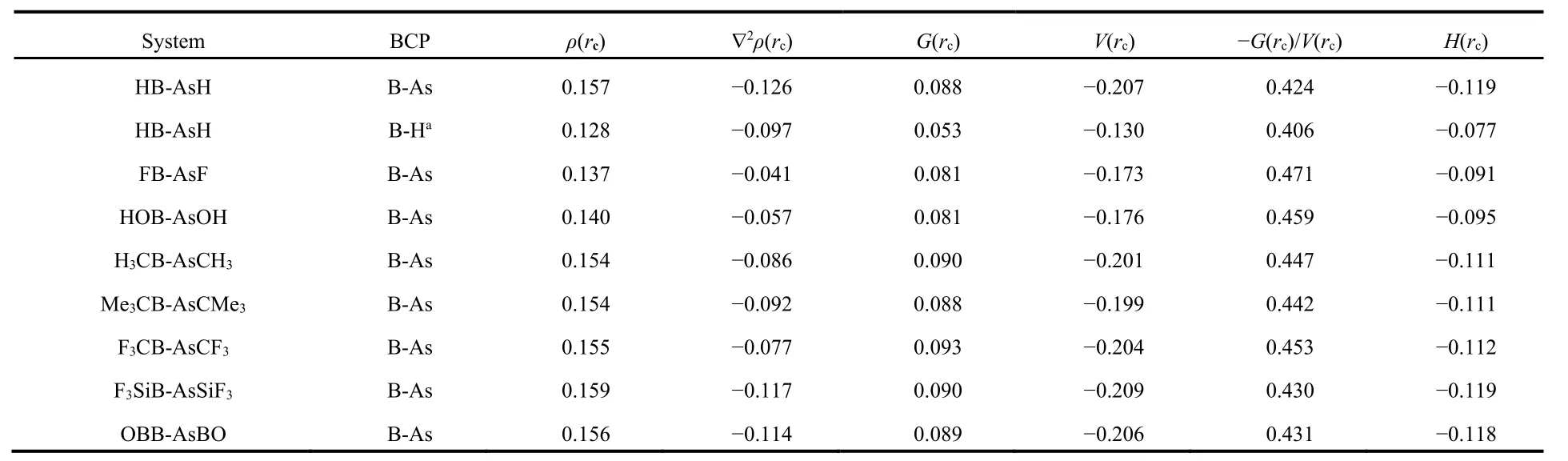
Table 3 Electron density descriptors (in a.u.) at the bond critical points (BCP) in between B and As atoms in RB-AsR (R = H, F, OH, CH3, CMe3, CF3, SiF3 and BO) at M06-2X/def2-TZVP level.
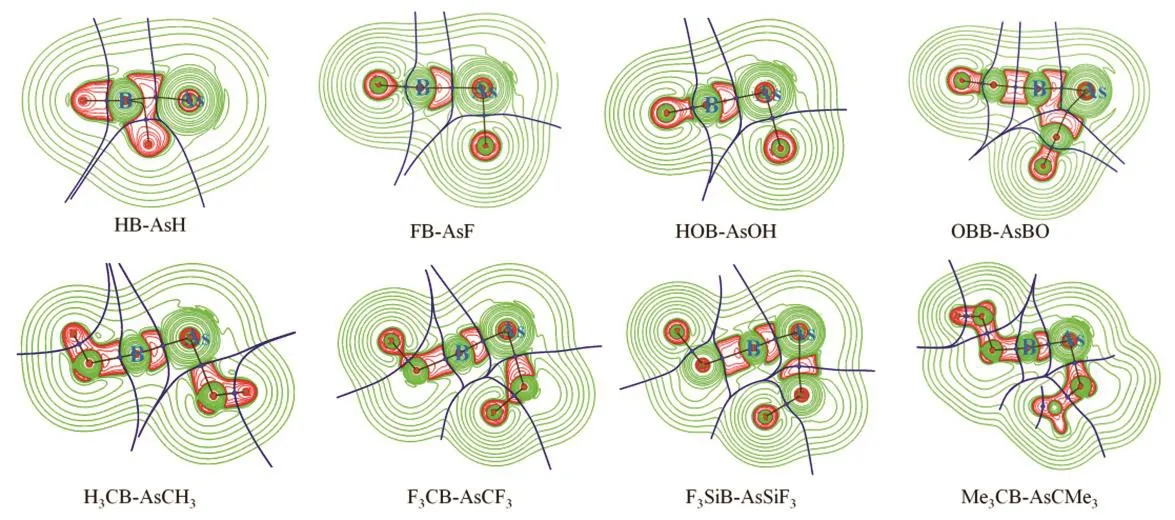
Fig.4 Plots of Laplacian of electron density (▽2ρ(r)) of RB-AsR (R = H, F, OH, CH3, CMe3, CF3, SiF3 and BO) systems at M06-2X/def2-TZVP level.
4.3 Reactivity analysis
In order to get insights into the reactivity on different sites of these systems and to examine the utility of the multiphilic descriptor Δωkwe have calculated local reactivity as well as selectivity descriptors using natural population analyis scheme.The results are given in Table 4. Here, we have taken RB-AsR,R'B-AsR and RB-AsR' systems, where the R and R' groups include H, electron donating CH3, CMe3, OH and electron withdrawing F, CF3, SiF3, BO. Although, the Fukui function descriptor is very useful in determining the preferable site for nucleophilic as well as electrophilic attacks in these systems,but it is unable to predict this when different systems are compared. Among these studied systems in HB-AsH,OHB-AsOH, OBB-AsBO and H3CB-AsBO, the f+as well as f?on As centre is greater than that in B centre. Also, the values of f+on B are slightly greater than As centres in most cases and thus making it difficult to get a clear decision on electrophilic behavior of these sites. To overcome this problem another reactivity descriptor Δω, called multiphilic descriptor is proposed. The advantage of Δω in comparison to Δf is that it contains all the global information along with local properties.Since, Δω is positive on B centre and negative on As centre in almost all of the systems studied showing that former is preferable for nucleophilic attack and the latter for electrophilic attack and this is expected because of the electron deficiency at the B centre in all the systems. Although, in OBB-AsBO system both centres contain negative Δω, the magnitude on As centre is much greater than that on B centre making the As centre more prone to nucleophilic attack. On an average an electron withdrawing group on the B centre improves the capability of nucleophilic attack whereas the propensity of the electrophilic attack improves on the As centre in case it is attached to an electron donating group. The trend, however, is not universal due to the known inadequacy of the population analysis scheme used.
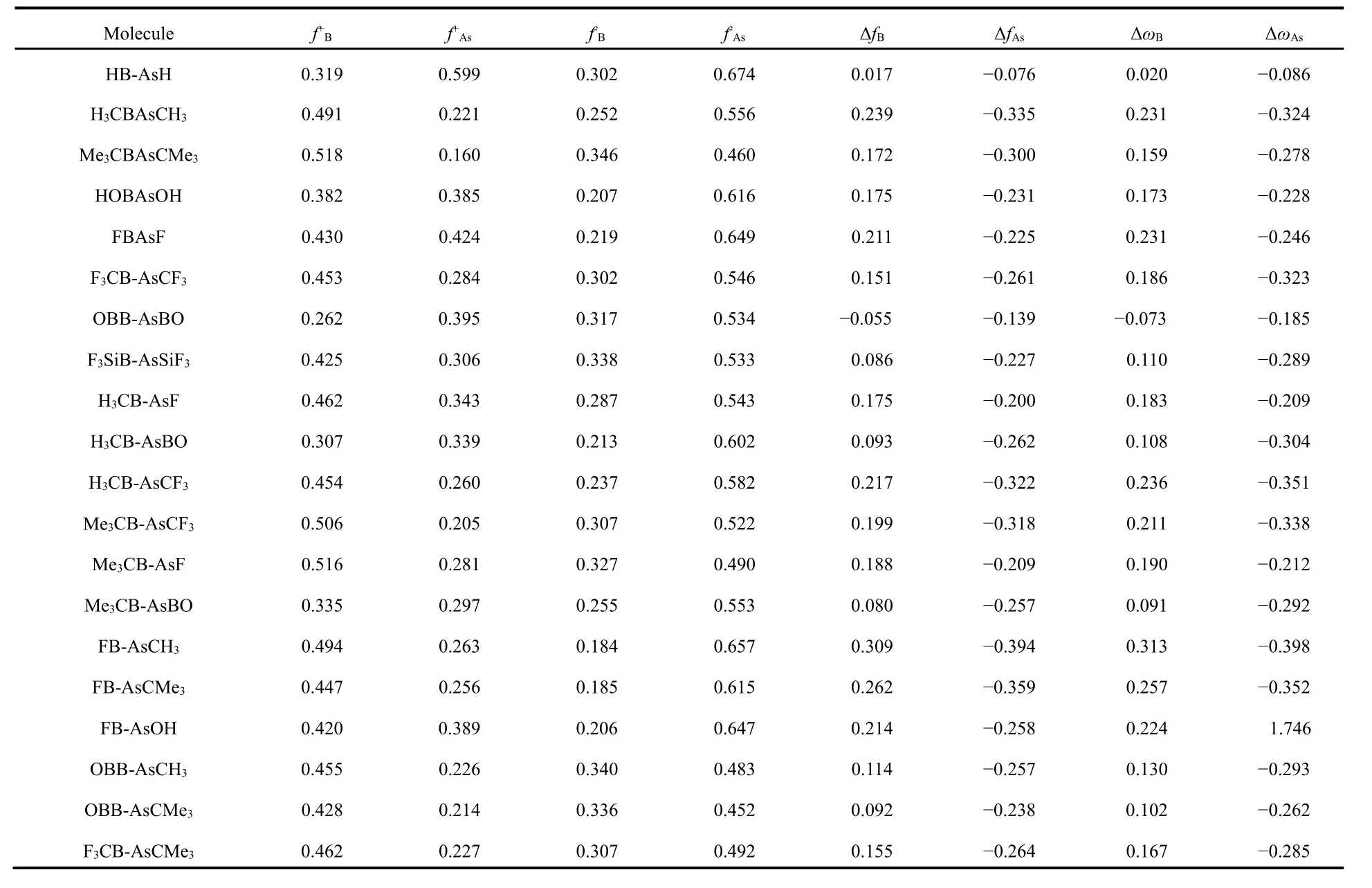
Table 4 Calculated local reactivity properties of some selected molecules calculated at the M06-2X/def2-TZVP level.
5 Conclusions
The most significant result obtained in this study reveals that all the RB-AsR systems adopt a bent geometry with∠B―As―R ≈ 90° or less and ∠R―B―As ≈ 180°. The reason behind these bent geometries as schematically presented in Fig.1 is analyzed. It is also shown that Scheme-II is favorable for a strong B―As bond formation, which is reflected in the values of the corresponding HOMO-LUMO gap. The computed WBI values show that B=As double bond is present in most of the cases. However, in some cases a weak triple bond is present, as suggested by the WBI value of say, B―As bond to be 2.25 in F3SiB-AsSiF3 system. The electron density analysis highlights the nature of the bonds present in these systems. We also calculated the multiphilic descriptors on B and As sites in RB-AsR systems, which give positive values on B centers and negative values on As centers in almost all of the systems highlighting the fact that the former is preferable for nucleophilic attack and latter is apt for electrophilic attack.
This article is dedicated to the memory of the late Professor Robert G. Parr, a great scientist and an excellent human being.PKC would like to thank Professor Shubin Liu for kindly inviting him to contribute an article for this special issue.
(2) Bino, A.; Ardon, M.; Shirman, E. Science 2005, 308, 234.doi: 10.1126/science.1109965
(3) Seidu, I.; Seth, M.; Ziegler, T. Inorg. Chem. 2013, 52, 8378.doi: 10.1021/ic401149h
(4) Danovich, D.; Bino, A.; Shaik, S. J. Phys. Chem. Lett. 2013, 4, 58.doi: 10.1021/jz3016765
(5) Lein, M.; Krapp, A.; Frenking, G.; J. Am. Chem. Soc. 2005, 127,6290. doi: 10.1021/ja042295c
(6) Sekiguchi, A.; Kinjo, R.; Ichinohe, M. Science 2004, 305, 1755.doi: 10.1126/science.1102209
(7) Sasamori, T.; Hironaka, K.; Sugiyama, T.; Takagi, N.; Nagase, S.;Hosoi, Y.; Furukawa, Y.; Tokitoh, N. J. Am. Chem. Soc. 2008, 130,13856. doi: 10.1021/ja8061002
(8) Stender, M.; Phillips, A. D.; Wright, R. J.; Power, P. P. Angew.Chem., Int. Ed. 2002, 41, 1785. doi: 10.1002/1521-3773(20020517)41:10<1785::AID-ANIE1785>3.0.CO;2-6
(9) Sugiyama, Y.; Sasamori, T.; Hosoi, Y.; Furukawa, Y.; Takagi, N.;Nagase, S.; Tokitoh, N. J. Am. Chem. Soc. 2006, 128, 1023.doi: 10.1021/ja057205y
(10) Phillips, A. D.; Wright, R. J.; Olmstead, M. M.; Power, P. P. J.Am. Chem. Soc. 2002, 124, 5930. doi: 10.1021/ja0257164
(11) Pu, L.; Twamley, B.; Power, P. P. J. Am. Chem. Soc. 2000,122, 3524. doi: 10.1021/ja993346m
(12) Wu, P. C.; Su, M. D. Dalton Trans. 2011, 40, 4253.doi: 10.1039/C0DT00800A
(13) Wu, P. C.; Su, M. D. Inorg. Chem. 2011, 50, 6814.doi: 10.1021/ic200930v
(14) Paetzold, P. Adv. Inorg. Chem. 1987, 31, 123.doi: 10.1016/S0898-8838(08)60223-8
(15) Petrie, M. A.; Shoner, S. C.; Dias, H. V. R.; Power, P. P.Angew. Chem., Int. Ed. Engl. 1990, 29, 1033.doi: 10.1002/anie.199010331
(16) Rivard, E.; Merrill, W. A.; Fettinger, J. C.; Power, P. P. Chem.Commun. 2006, 3800. doi: 10.1039/b609748k
(17) Mardones, M. A.; Cowley, A. H.; Contreras, L.; Jones, R. A.J. Organomet. Chem. 1993, 455.doi: 10.1016/0022-328X(93)80411-4
(18) Kerins, M. C.; Fitzpatrick N. J.; Nguyen, M. T. Polyhedron 1989, 8, 969. doi: 10.1016/S0277-5387(00)86453-0
(19) Watts, J. D.; Zant, L. C. V. Chem. Phys. Lett. 1996, 251, 119.(20) Lu, J. S.; Yanga, M. C.; Su, M. D. PhysChemChemPhys 2017,19, 8026. doi: 10.1039/c7cp00421d
(21) Lu, J. S.; Su, S. H.; Yanga, M. C.; Wen, X. T.; Xie, J. Z.; Su,M. D. Organometallics 2016, 35, 3924.doi: 10.1021/acs.organomet.6b00659
(22) Lu, J. S.; Yanga, M. C.; Su, M. D. ACS Omega 2017, 2, 1172.doi: 10.1021/acsomega.7b00113
(23) Chattaraj, P. K.; Rivas, N. G.; Matus, M. H.; Galvan, M. J.Phys. Chem. A 2005, 109, 25. doi: 10.1021/jp045319a
(24) Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989.(25) Geerlings, P.; De Proft, F.; Langenaeker, W. Chem. Rev. 2003,103, 1793. doi: 10.1021/cr990029p
(26) Parr, R. G.; Von Szentpaly, L.; Liu, S. J. Am. Chem. Soc.1999, 121, 1922. doi: 10.1021/ja983494x
(27) Chattaraj, P. K.; Sarkar, U.; Roy, D. R. Chem. Rev. 2006, 106,2065. doi: 10.1021/cr078014b
(28) Parr, R. G.; Donnelly, R. A.; Levy, M.; Palke, W. E. J. Chem.Phys.1978, 68, 3801. doi: 10.1063/1.436185
(29) Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105,7512. doi: 10.1021/ja00364a005
(30) Chattaraj, P. K.; Parr, R. G. Density Functional Theory of Chemical Hardness in Chemical Hardness (Structure and Bonding) Vol. 80; Sen, K. D., Mingos, D. M. P. Eds.;Springer: Berlin, Germany, 1993.
(31) Chattaraj, P. K.; Giri, S.; Duley, S. J. Phys. Chem. Lett. 2010,1, 1064. doi: 10.1021/jz1001117
(32) Sanderson, R. T. Science 1951, 114, 670.doi: 10.1126/science.114.2973.670
(33) Datta, D. J. Phys. Chem. 1986, 90, 4216.doi: 10.1021/j100408a076
(34) Szentpály, L. V. J. Phys. Chem. A 2015, 119, 1715.doi: 10.1021/jp5084345
(35) Szentpály, L. V. J. Mol. Model. 2017, 23, 217.doi: 10.1007/s00894-017-3383-z
(36) Posthumus, K. Rec. Trav. Chim. 1933, 52, 25; 1933, 53, 308.(37) Parr, R. G.; Yang, W. J. Am. Chem. Soc. 1984, 106, 4049.doi: 10.1021/ja00326a036
(38) Yang, W.; Mortier, W. J. J. Am. Chem. Soc. 1986, 108, 5708.doi: 10.1021/ja00279a008
(39) Chattaraj, P. K.; Maiti, B.; Sarkar, U. J. Phys. Chem. A 2003,107, 4973. doi: 10.1021/jp034707u
(40) Roy, D. R.; Parthasarathi, R.; Padmanabhan, J.; Sarkar, U.;Subramanian, V.; Chattaraj, P. K. J. Phys. Chem. A 2006, 110,1084. doi: 10.1021/jp053641v
(41) Morell, C.; Grand, A.; Toro-Labbe′, A. J. Phys. Chem. A 2005, 109, 205. doi: 10.1021/jp046577a
(42) Morell, C.; Grand, A.; Toro-Labbe, A. Chem. Phys. Lett.2006, 425, 342. doi: 10.1016/j.cplett.2006.05.003
(43) Padmanabhan, J.; Parthasarathi, R.; Elango, M.;Subramanian, V.; Krishnamoorthy, B. S.; Gutierrez-Oliva, S.;Toro-Labbé, A.; Roy, D. R.; Chattaraj, P. K. J. Phys. Chem. A 2007, 111, 37. doi: 10.1021/jp0718909
(44) Chattaraj, P. K.; Chakraborty, A.; Giri, S. J. Phys. Chem. A.2009, 113, 10068. doi: 10.1021/jp904674x
(45) Zhao, Y.; Truhlar, D. G. Theor. Chem. Account. 2006, 120,215. doi: 10.1007/s00214-007-0310-x
(46) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 09, Revision C.01; Gaussian Inc.: Wallingford CT, 2010.
(47) Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988,88, 899. doi: 10.1021/cr00088a005
(48) Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys.1985, 83, 735. doi: 10.1063/1.449486
(49) Wiberg, K. B. Tetrahedron 1968, 24, 1083.doi: 10.1016/0040-4020(68)88057-3
(50) Bader, R. F. W. Atoms in Molecules: A Quantum Theory;Clarendon Press: Oxford, UK, 1990.
(51) Lu, T.; Chen, F. W. J. Comput. Chem. 2012, 33, 580.doi: 10.1002/jcc.22885
(52) Pyykko, P.; Desclaux, J. P. Acc. Chem. Res. 1979, 12, 276.doi: 10.1021/ar50140a002
(53) Pyykko, P. Chem. Rev. 1988, 88, 563.doi: 10.1021/cr00085a006
