微通道費-托合成反應規律及本征動力學
2018-03-05 05:46:14侯朝鵬胡志海
石油學報(石油加工) 2018年1期
顧 暢, 侯朝鵬, 徐 潤, 胡志海
(中國石化 石油化工科學研究院, 北京 100083)
費-托合成是指在一定溫度、壓力下將合成氣(CO和H2)轉化為烴類及含氧化合物的方法,該技術目前已經成為煤炭、天然氣、生物質等含碳資源清潔優化利用的重要途徑[1]。然而費-托合成是強放熱反應,擴散限制影響也較大[2],因此反應器體系傳熱傳質效率對于裝置穩定運行、延長催化劑壽命、改善產物分布具有重要意義。微通道反應器是指反應空間至少一個維度的尺寸小于1 mm的反應系統[3],其傳熱、傳質效率和表面積都大大增加,反應控制更加精細。將微通道反應器應用于費-托合成,能解決傳統固定床反應器傳熱、傳質差以及漿態床反應器固、液分離的問題,是費-托合成一個重要的發展方向[4-6]。
對于目前已工業應用的費-托合成反應器,有關催化劑和工藝的研究報道較多[7-9]。然而由于在傳質和傳熱上的優勢,微通道反應器費-托合成不僅催化活性大大提高,而且反應規律相比于傳統反應器也存在諸多不同。例如,Cao等[10]開展了狹縫式微通道費-托合成的研究,其測試的鈷基催化劑在235℃、60000 h-1條件下,轉化率達到63%,表現較高的催化活性;增加壓力或降低n(H2)/n(CO)可以明顯抑制CH4生成反應,顯著提高鏈增長因子。Almeida等[3,11]考察了在微通道反應模塊上空速及壓力對于費-托合成活性和選擇性的影響,CO分壓增高會降低反應速率;隨著空速的增加,CH4選擇性顯著降低,鏈增長因子有所增加;而壓力對于活性和選擇性的影響均較小。Leviness等[12]在微通道體系中開展了催化劑長周期穩定性研究。得益于較高的傳熱效率,微通道反應器中可以使用更高活性催化劑,且催化劑在單程轉化率超過80%、出口pH2O/pH2達到7~8的條件下仍保持較好的穩定性,且可以多次重復再生。
動力學研究可以為費-托合成反應器放大和工藝條件優化提供基礎。早期的研究在Co/Al2O3催化劑上建立了冪函數型CO消耗速率方程,將CO消耗速率與H2分壓和CO分壓關聯起來[13-14]。近些年的研究通過假設基元反應和速率控制步驟建立了LHHW(Langmuir-Hinshelwood-Hougen-Watson)型CO消耗速率方程[15-17]。Visconti等[18]在粒徑75~106 μm的Co/Al2O3催化劑上進行了詳細動力學模型的研究,模型能夠對反應物轉化率和 C1~C50的烴類產物分布進行準確預測,并且能夠解釋產物分布偏離ASF模型的現象。Almeida等[11]基于微通道反應體系進行了詳細動力學模型的研究。模型并未涉及鏈增長因子α,僅僅是溫度和反應物分壓的函數。在轉化率小于 50%的條件下,擴散限制較弱,模型適用性較好。因此微通道反應體系較高的空速有利于提高動力學模型的準確性。
在微通道反應體系中系統地研究工藝參數對于反應的影響,可以加深對反應過程的認識,并且有助于微通道費-托合成工藝的開發。然而目前文獻報道中尚無系統全面的微通道費-托合成工藝研究。另一方面,動力學研究不僅可以對反應速率進行預測,而且可以進行定量計算,為反應器的結構設計和優化提供理論指導。筆者在微通道反應體系中,系統地考察了溫度、壓力、體積空速(以下簡稱空速)、n(H2)/n(CO) 4個主要工藝參數對于鈷基催化劑費-托合成反應活性和選擇性的影響,并擬合得到本征動力學方程,從而為費-托合成反應器的模擬優化奠定基礎。
1 實驗部分
1.1 原料氣及裝置流程
原料氣為n(H2)/n(CO)為 2.0和1.0的合成氣,其中包含一定量的N2作為內標氣。原料氣經過質量流量計后進入凈化罐,脫除微量的雜質。凈化后的原料氣進入反應器,生成的氣-液混合相流入熱阱,分離出高沸點的產物。分離后的氣相進入冷阱,進一步將水和油等低沸點產物分離。尾氣使用Varian的GC4900型氣相色譜進行組成分析。
1.2 催化劑裝填及還原
催化劑為中國石化石油化工科學研究院研制的Co/Al2O3費-托合成催化劑,裝填于徑向尺寸約0.5 mm的環形狹縫中,裝填量為0.36 g,裝填體積約為0.25 mL。使用相同粒徑石英砂稀釋8倍,以減少床層內部溫差,達到均溫效果。催化劑使用H2進行還原,空速為10000 h-1,升溫速率為5℃/min,還原溫度為400℃,還原時間為4 h。
2 結果與討論
2.1 催化劑粒度和反應條件對Co/Al2O3催化劑費-托合成反應活性及選擇性的影響
2.1.1 催化劑粒度的影響
進行本征動力學實驗,首先需要消除內外擴散的影響,使催化劑活性位上的原料組成接近體相組成,并且單程轉化率不能過高。
相同催化劑裝量下,空速大小決定催化劑表面氣流的線速度和反應的單程轉化率;而內擴散主要受催化劑粒徑影響。因此為了消除內外擴散和轉化率對反應的影響,本實驗將催化劑篩分成50~75 μm、75~100 μm、100~125 μm、125~150 μm 4個粒徑范圍,并在230℃(動力學數據點采集實驗的最高溫度)、n(H2)/n(CO)為2.0的條件下,考察空速以及催化劑粒徑對于CO消耗速率及產物選擇性的影響,從而確定能夠消除內、外擴散影響的實驗條件。實驗結果如表1所示。
由表1可見,當空速為20000 h-1時,CO消耗速率明顯低于高空速條件的結果,表明此時轉化率過高,床層后段反應物分壓過低。而對于不同粒徑的Co/Al2O3催化劑,空速達到40000 h-1后活性和選擇性均變化不大,說明空速高于40000 h-1時,外擴散影響得到基本排除。
內擴散方面,對于空速為60000 h-1的實驗結果,在Co/Al2O3催化劑粒徑增加到100~125 μm時,CH4選擇性略有增加,C5+選擇性小幅下降,表明開始出現內擴散影響。在Co/Al2O3催化劑粒徑為125~150 μm時,CH4選擇性繼續增加,出現較顯著的內擴散影響。因此可以認為Co/Al2O3催化劑粒徑在100 μm以內時,內擴散影響得到基本排除。
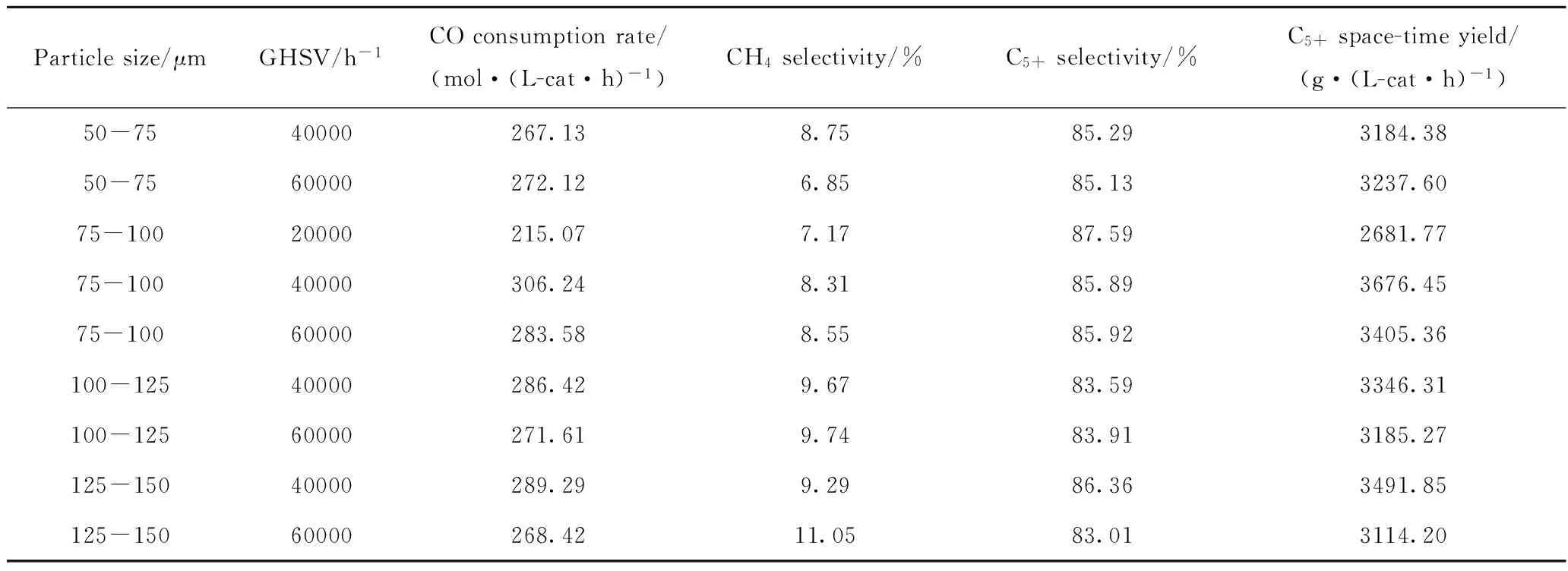
表1 Co/Al2O3催化劑粒徑及空速對費-托合成反應活性及選擇性的影響Table 1 Effect of catalyst particle size and space velocity on activity and selectivity of Fischer-Tropsch synthesis over Co/Al2O3 catalyst
Reaction conditions:T=230℃;p=2.5 MPa;n(H2)/n(CO)=2.0
2.1.2 空速的影響
在2.5 MPa、n(H2)/n(CO)=2.0的條件下,考察不同反應溫度下空速對于微通道Co/Al2O3催化劑費-托合成反應活性及選擇性的影響,結果如表2 所示。由于空速改變,CO轉化率必然隨之改變,因此以CO消耗速率來表示催化劑的活性。
由表2可知,220℃條件下,空速為20000 h-1和40000 h-1時,CO消耗速率無明顯變化。230℃條件下,隨著空速的增加,CO消耗速率先增加,在空速高于40000 h-1時,活性進入平臺區,不再隨空速增加而增加。選擇性方面, 230℃條件下,隨著空速增加,CH4選擇性稍有增加,C5+選擇性小幅下降,空速高于40000 h-1時,選擇性不再變化。這一規律與文獻[9]報道中固定床費-托合成的反應結果明顯不同。
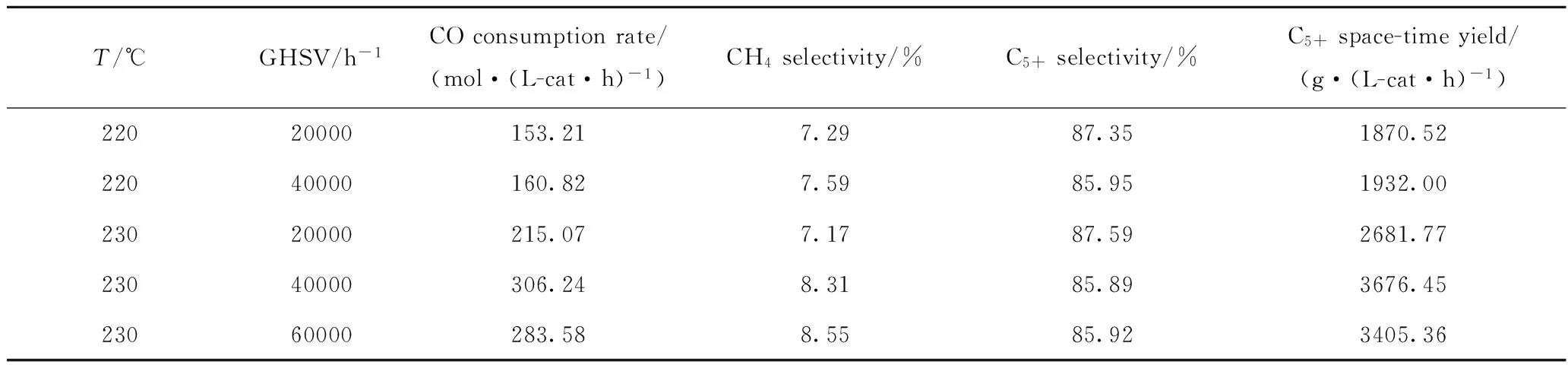
表2 不同溫度下Co/Al2O3催化劑上體積空速對費-托合成反應活性和選擇性的影響Table 2 Effect of volume space velocity on activity and selectivity of Fischer-Tropsch synthesis over Co/Al2O3 catalyst at different temperatures
Reaction conditions:p=2.5 MPa;n(H2)/n(CO)=2.0
2.1.3 溫度的影響
在2.5 MPa、空速20000~80000 h-1、n(H2)/n(CO)=2.0的條件下,考察反應溫度對微通道Co/Al2O3催化劑費-托合成性能的影響,以CO消耗速率來表示催化劑的活性,結果如圖1所示。由圖1可見,CO消耗速率隨溫度呈明顯的指數式變化。擬合得到對數形式的阿倫尼烏斯方程如式(1)所示。
lnrCO=36.31547-15494.13549/T
(1)
由擬合直線的斜率求得表觀活化能為128.8kJ/mol,明顯高于文獻[16-17,19-20]報道中80~120kJ/mol的水平。費-托合成反應生成的液態產物會包覆并填充催化劑,阻礙反應物的吸附以及產物的擴散,因此反應的表觀活化能也會受到擴散阻滯作用的影響。Kapteijn等[21]的研究中,催化劑涂層厚度為110μm和50μm時,活化能分別為45kJ/mol和60kJ/mol,而當涂層厚度達到30μm時,活化能進一步增加至115kJ/mol。這表明擴散限制的存在會顯著降低反應的表觀活化能。因此在微通道反應體系中,由于內外擴散影響大幅削弱,費-托合成反應的表觀活化能較高,更加接近于其本征活化能。同時微通道反應體系傳熱效率高,溫度控制更加精細,有利于溫度敏感性高的反應的工藝開發。
由圖1還看到,隨著溫度升高,CH4的選擇性從4.0%增加到10.6%,C5+選擇性則相應地從89.0%下降到81.5%,這是因為鏈增長反應的活化能低于鏈終止反應的活化能,升高溫度對于鏈終止反應的促進效果更加顯著。
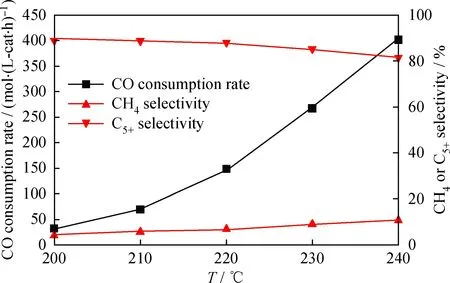
圖1 反應溫度對Co/Al2O3催化劑費-托合成 反應活性和選擇性的影響Fig.1 Effect of temperature on activity and selectivities of Fischer-Tropsch synthesis Reaction conditions: p=2.5 MPa; n(H2)/n(CO)=2.0; GHSV=20000-80000 h-1
2.1.4 壓力的影響
在220℃、空速40000 h-1、n(H2)/n(CO)=2.0的條件下,考察壓力對于Co/Al2O3催化劑費-托合成反應活性及選擇性的影響,結果如圖2所示。由圖2看到,隨著壓力增加,CO轉化率近似于線性增加;而壓力大于2.0 MPa后,該規律逐漸轉變為對數形式變化;在4.0 MPa以后,壓力的增加對于催化劑活性的提升趨緩。這是因為增加壓力使H2和CO的吸附平衡正向移動,由于H2分壓對活性C物種加氫反應促進作用明顯,因此反應速率有所增加。而壓力達到4.0 MPa后,催化劑活性位表面已趨近于飽和吸附,因此反應速率增速減緩直至達到平臺。由圖2還看到,隨著壓力增加,CH4選擇性從7.96%降到6.29%,C5+選擇性則是從84.30%增加到88.96%。總體來說,壓力對于選擇性的影響較小,這一規律與文獻[9]報道中固定床反應體系的實驗結果存在較為明顯的差異,而與Almeida等[3]和Myrstad等[22]在微通道反應體系中得到的結果較為接近。
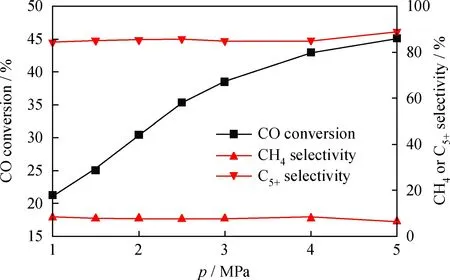
圖2 反應壓力對Co/Al2O3催化劑費-托合成 反應活性和選擇性的影響Fig.2 Effect of pressure on activity and selectivities of Fischer-Tropsch synthesis Reaction conditions: T=220℃; GHSV=40000 h-1; n(H2)/n(CO)=2.0
2.1.5n(H2)/n(CO)的影響
在220℃、2.5 MPa、空速40000 h-1的條件下,改變原料氣n(H2)/n(CO),考察n(H2)/n(CO)對于Co/Al2O3催化劑費-托合成反應活性及選擇性的影響,結果如圖3所示。
由圖3可見,隨著n(H2)/n(CO)從1.0增加至3.0,CO轉化率從19.10%顯著增加到63.33%,H2的轉化率也有小幅提高。與圖2結果對比可以發現,反應速率對n(H2)/n(CO)更敏感,而對總壓敏感性較小。選擇性方面,隨著n(H2)/n(CO)增加,CH4選擇性迅速從3.58%增加至8.08%,C5+選擇性相應下降,產物中乙烯與乙烷的摩爾比從0.38大幅降至0.03。可能的原因是n(H2)/n(CO)的增加,使得原料氣中CO分壓顯著下降,從而降低了Co/Al2O3催化劑活性位上活性C物種的濃度,阻礙了鏈增長反應;并且H2分壓的提高,增加了活性C物種加氫速率,從而使CH4的生成反應以及烯烴加氫的二次反應加強。雖然C5+產物的選擇性有所下降,但由于CO消耗速率大幅提升,C5+時空收率也由1.83 kg/(L-cat·h)增加到3.10 kg/(L-cat·h)。因此可以適當提高原料氣n(H2)/n(CO),以提高反應速率,獲得更多液態烴類產物。
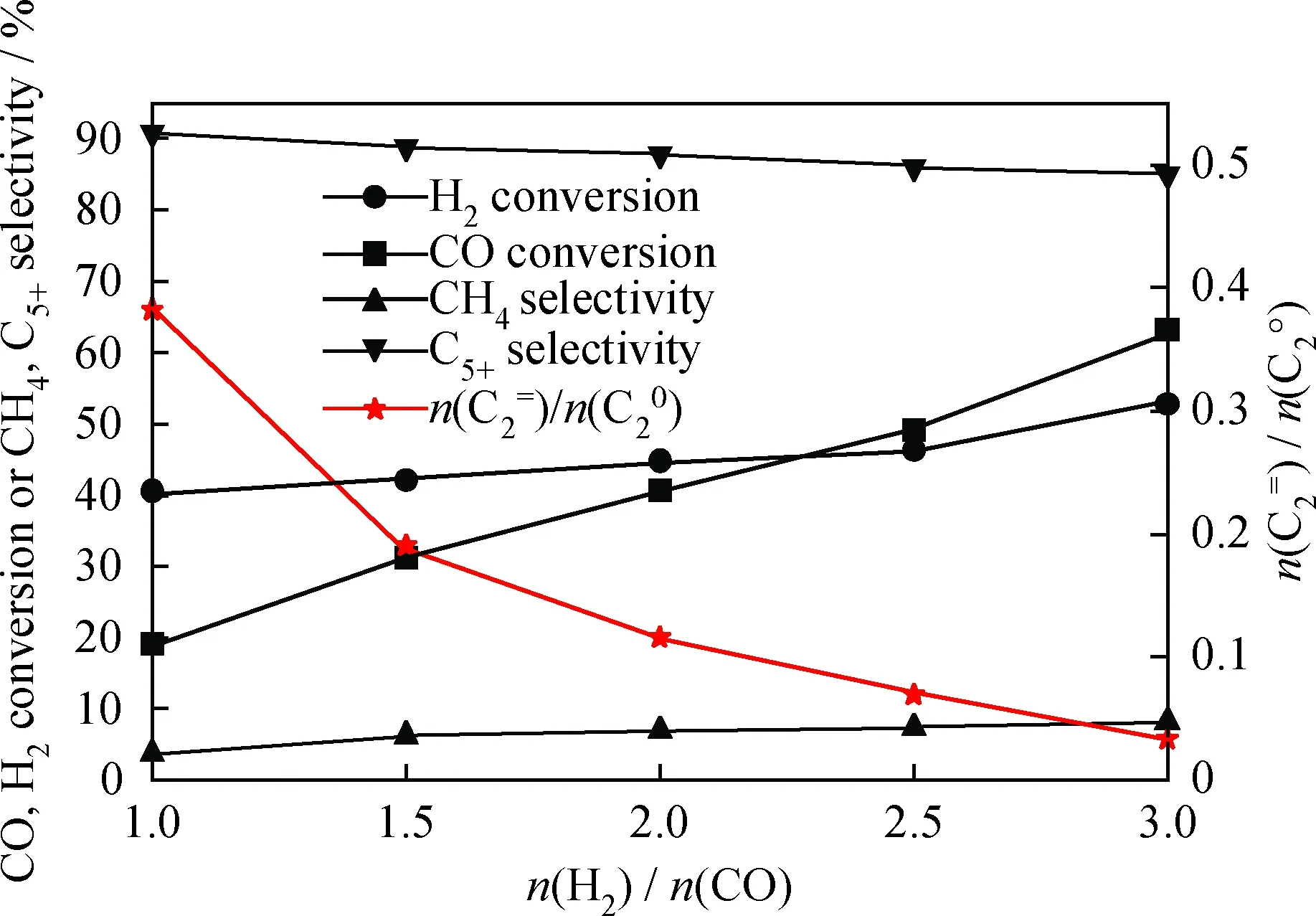
圖3 n(H2)/n(CO)對Co/Al2O3催化劑費-托合成 反應活性和選擇性的影響Fig.3 Effect of n(H2)/n(CO) on activity and selectivities of Fischer-Tropsch synthesis Reaction conditions: T=220℃; p=2.5 MPa; GHSV=40000 h-1
2.2 Co/Al2O3催化劑費-托合成反應本征動力學模型選擇和參數估計
本征動力學實驗固定空速為40000 h-1,以溫度、壓力、n(H2)/n(CO)為變量,每個變量取3個水平,采用全面實驗方案共獲得27個數據點。剔除轉化率過高和偏差較大的數據點后共獲得25個有效數據點,結果見表3。
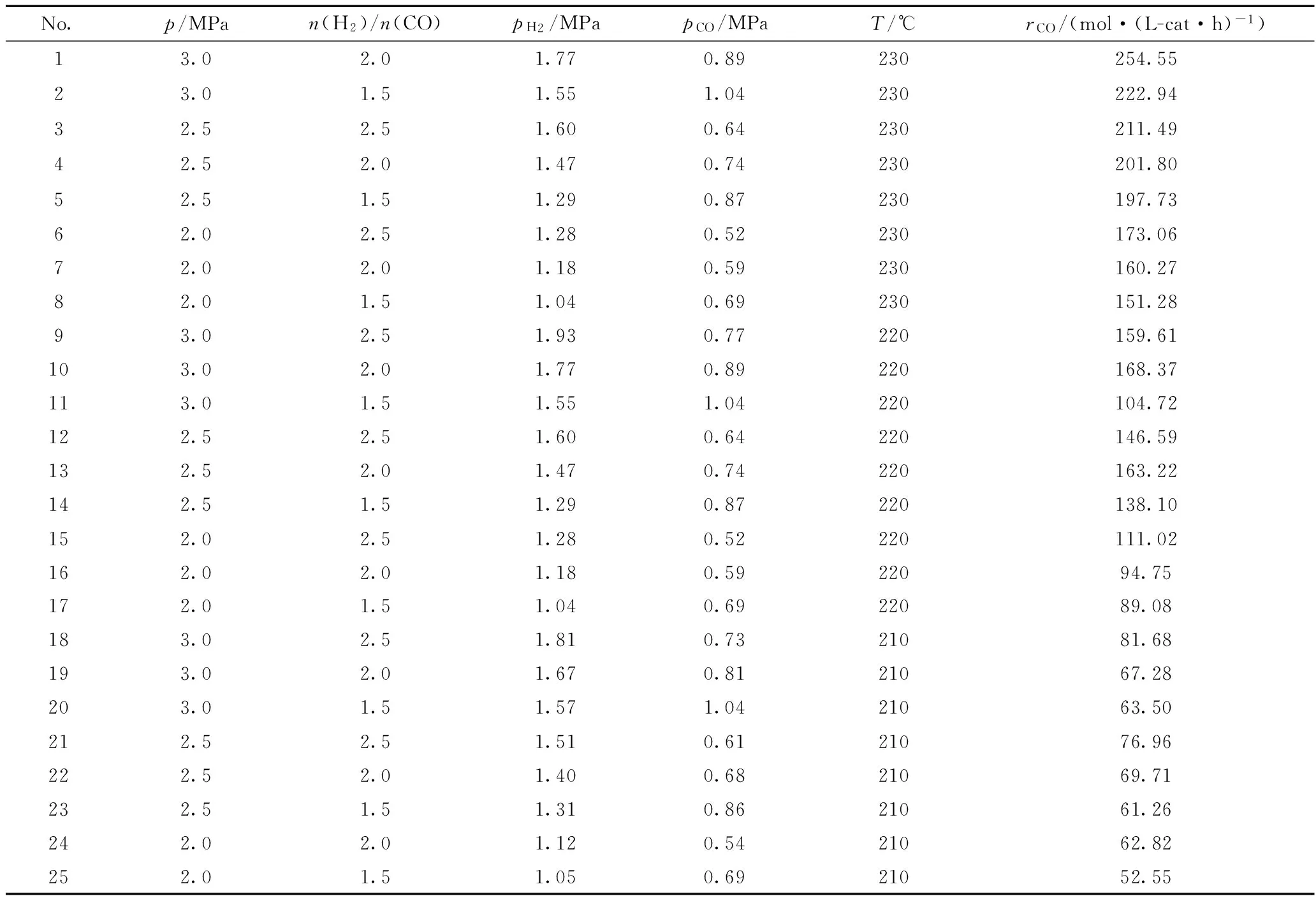
表3 Co/Al2O3催化劑費-托合成反應動力學實驗數據Table 3 Kinetic experimental results of Fischer-Tropsch synthesis over Co/Al2O3 catalyst
費-托合成動力學模型可以分為CO消耗速率模型以及產物生成速率模型。CO消耗速率模型又可以分為冪函數型和通過反應機理的假設以及速率控制步驟的選擇推導得出的LHHW型。詳細的產物生成速率模型需要基于產物分布模型推導而出,模型的建立較為復雜[9,16-17,23-24],因此本實驗目標為建立CO消耗速率模型。目前已有較多有關LHHW型CO消耗速率方程的研究,從中選擇與本實驗體系相近的研究中[9,16]推導的LHHW型方程以及簡單冪函數型方程為備選模型。在Python軟件中,采用最小化算法進行參數優化,以CO消耗速率方程的計算值和實驗值的相對殘差絕對值的平均值(MARR值)為目標函數,如式(2)所示。
(2)
式(2)中,Nexp為數據點個數;rexp為CO消耗速率實驗值,mol·(L-cat·h)-1;rcal為CO消耗速率計算值,mol·(L-cat·h)-1。
表4為備選CO消耗速率模型及各模型擬合結果的MARR值。由表4結果可知,模型5的MARR值最小,擬合效果最好。而模型1的簡單冪函數型模型,方程形式簡單,并且MARR值較小,同樣具有較好的擬合效果。因此綜合考慮模型的易用性和擬合效果,最終選取的CO消耗速率模型見式(3)。
(3)
式(3)中,rCO為CO消耗速率,mol·(L-cat·h)-1;k為反應速率常數,mol·(L·h)-1·MPa-(a+b);a、b分別為H2、CO的反應級數。
模型擬合得到的參數如表5所示。因為溫度、壓力等工藝參數取值范圍存在較大不同,所以擬合得到的活化能與前述實驗結果也有一定的差別,但仍然處于文獻報道結果中較高的水平。對所建模型進行相對誤差分析,平均絕對相對殘差MARR為7.77%,擬合效果較好。實驗值與計算值的相對偏差如圖4所示。由圖4可見,80%數據點的模型計算值和實驗值的相對偏差在15%以內,表明所建CO消耗速率模型具有較好的適用性。其中參數a、b的值分別為0.94和-0.04,表明提高H2分壓能夠有效提高反應速率,這與前述實驗結果相符。并且模型可進一步近似簡化為溫度項和H2分壓的一次函數項,這將使模型的應用更加簡便。

表4 Co/Al2O3催化劑費-托合成反應中CO消耗速率模型及擬合結果的相對殘差絕對值的平均值(MARR)Table 4 CO consumption rate expressions and mean absolute relative residuals(MARR) of fitting results of Fischer-Tropsch synthesis over Co/Al2O3 catalyst
a,b,c,dare kinetic constants in expression 2-11.
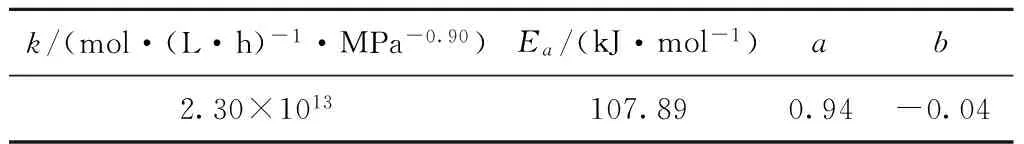
表5 Co/Al2O3催化劑費-托合成反應 動力學模型參數擬合結果Table 5 Estimated parameters of the kinetic model of Fischer-Tropsch synthesis over Co/Al2O3 catalyst
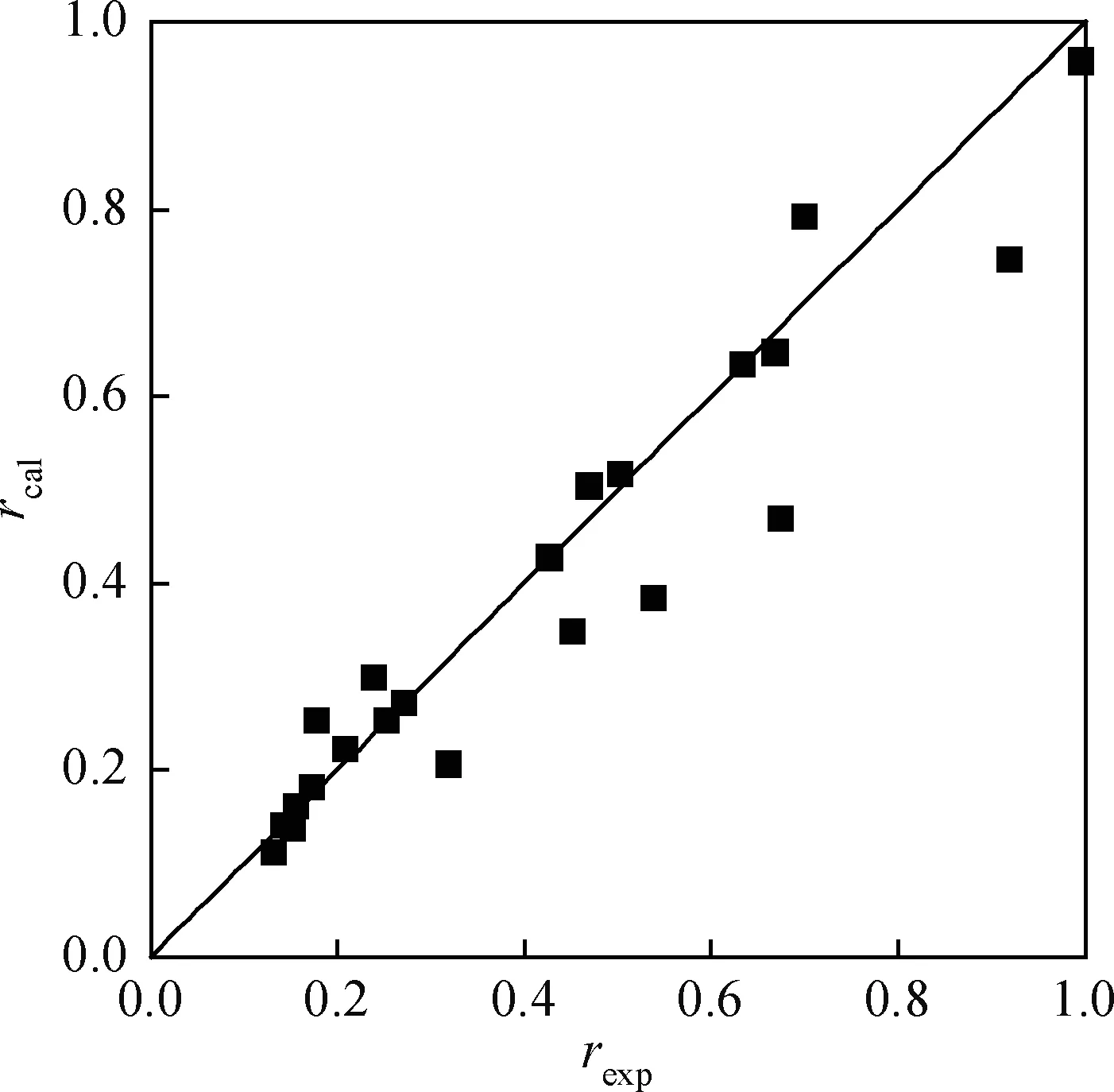
圖4 Co/Al2O3催化劑費-托合成反應中CO消耗 速率實驗值與計算值相對偏差Fig.4 Comparison between experimental and calculated values of CO consumption rate of Fischer-Tropsch synthesis over Co/Al2O3 catalyst rexp—Experimental value; rcal—Calculated value
3 結 論
(1)在微通道Co/Al2O3催化劑費-托合成反應體系中,在Co/Al2O3催化劑粒徑小于100 μm、空速超過40000 h-1的條件下,內外擴散影響大幅削弱,Co/Al2O3催化劑表現出接近于本征狀態的活性和選擇性,反應規律相比于傳統反應器中的實驗結果存在明顯的差異。
(2)在微通道Co/Al2O3催化劑費-托合成反應體系中,由于擴散影響大幅降低,Co/Al2O3催化劑的表觀活化能為128 kJ/mol,處于文獻報道中較高的水平。隨著空速的增加,CO消耗速率先增加后進入平臺區;空速對于選擇性的影響則十分微弱。增加壓力,反應活性先有較為明顯的提升,壓力達到4 MPa后,活性進入平臺區,而選擇性變化不明顯。n(H2)/n(CO)直接決定催化劑活性位表面的H2和CO濃度比例,對活性和選擇性影響較大,在1.0~3.0的范圍內,增大n(H2)/n(CO)能夠提高原料氣的轉化率和C5+的時空收率。
(3)綜合考慮模型的易用性和擬合效果,選擇建立了微通道Co/Al2O3催化劑費-托合成反應體系的冪函數型CO消耗速率模型,表達式為:
模型平均相對偏差為7.77%,80%的數據點的計算值和實驗值的相對偏差在15%以內,擬合效果較好。并且由于CO分壓對于反應活性影響較小,模型可簡化為溫度項和H2分壓的一次函數項,進一步增強了模型的適用性。模型為Co/Al2O3催化劑費-托合成反應器數值模擬研究奠定了基礎,為工業化應用提供了理論參考。
[1] 代小平, 余長春, 沈師孔. 費-托合成制液態烴研究進展[J].化學進展, 2000, 12(3): 268-281. (DAI Xiaoping, YU Changchun, SHEN Shikong. Recent advances in the synthesis of liquid hydrocarbon via Fischer-Tropsch synthesis[J].Progress in Chemistry, 2000, 12(3): 268-281.)
[2] ALMEIDA L C, ECHAVE F J, SANZ O, et al. Fischer-Tropsch synthesis in microchannels[J].Chemical Engineering Journal, 2011, 167(2-3): 536-544.
[3] ALMEIDA L C, SANZ O, D’OLHABERRIAGUE J, et al. Microchannel reactor for Fischer-Tropsch synthesis: Adaptation of a commercial unit for testing microchannel blocks[J].Fuel, 2013, 110(4): 171-177.
[4] 穆金霞, 殷學鋒. 微通道反應器在合成反應中的應用[J].化學進展, 2008, 20(1): 60-75. (MU Jinxia, YIN Xuefeng. Application of microfluidic reactors on synthesis reactions[J].Progress in Chemistry, 2008, 20(1): 60-75.)
[5] GUETTEL R, TUREK T. Comparison of different reactor types for low temperature Fischer-Tropsch synthesis: A simulation study[J].Chemical Engineering Science, 2009, 64(5): 955-964.
[6] 徐潤, 胡志海, 聶紅. 微反應器技術在Fischer-Tropsch合成中的應用進展[J].化工進展, 2016, 35(3): 685-691.(XU Run, HU Zhihai, NIE Hong. Recent advances on Fischer-Tropsch synthesis in micro-reactor[J].Chemical Industry and Engineering Progress, 2016, 35(3): 685-691.)
[7] GERARD P, VANDER LAAN, BEENACKERS A A C M. Kinetics and selectivity of the Fischer-Tropsch synthesis: A literature review[J].Catalysis Reviews, 1999, 41(3-4): 255-318.
[8] VISCONTI C G, TRONCONI E, LIETTI L, et al. Development of a complete kinetic model for the Fischer-Tropsch synthesis over Co/Al2O3, catalysts[J].Chemical Engineering Science, 2007, 62(18): 5338-5343.
[9] 魯豐樂. 費托合成催化劑反應動力學研究與反應器數學模擬[D].上海: 華東理工大學, 2010.
[10] CAO C, HU J, LI S, et al. Intensified Fischer-Tropsch synthesis process with microchannel catalytic reactors[J].Catalysis Today, 2009, 140(3): 149-156.
[11] ALMEIDA L C, SANZ O, MERINO D, et al. Kinetic analysis and microstructured reactors modeling for the Fischer-Tropsch synthesis over a Co-Re/Al2O3catalyst[J].Catalysis Today, 2013, 215(41): 103-111.
[12] LEVINESS S, DESHMUKH S R, RICHARD L A, et al. Velocys Fischer-Tropsch synthesis technology—New advances on state-of-the-art[J].Topics in Catalysis, 2014, 57(6-9): 518-525.
[13] YANG C H, MASSOTH F E, OBLAD A G. Kinetics of CO+H2Reaction over Co-Cu-Al2O3Catalyst;Hydrocarbon Synthesis from Carbon Monoxide and Hydrogen[M].Washington D.C.: Am Chem Soc, 1979.
[14] RIBEIRO F H, WITTENAU A. Reproducibility of turnover rates in heterogeneous metal catalysis: Compilation of data and guidelines for data analysis[J].Catalysis Reviews, 1997, 39(1-2): 49-76.
[15] SARUP B,WOJCIECHOWSKI B W. Studies of the Fischer-Tropsch synthesis on a cobalt catalyst II Kinetics of carbon monoxide conversion to methane and to higher hydrocarbons[J].The Canadian Journal of Chemical Engineering, 1989, 67(1): 62-74.
[16] NIKPARSA P, MIRZAEI A A, ATASHI H. Effect of reaction conditions and kinetic study on the Fischer-Tropsch synthesis over fused Co-Ni/Al2O3catalyst[J].Journal of Fuel Chemistry & Technology, 2014, 42(6): 710-718.
[17] 常杰, 滕波濤, 白亮, 等. Co/ZrO2/SiO2催化劑費-托合成反應動力學研究Ⅱ動力學模型的構建和回歸[J].催化學報, 2005, 26(10): 859-868. (CHANG Jie, TENG Botao, BAI Liang, et al. Detailed kinetic study of Fischer-Tropsch synthesis on Co/ZrO2/SiO2catalyst Ⅱ Construction and regression of kinetic models[J].Chinese Journal of Catalysis, 2005, 26(10): 859-868.)
[18] VISCONTI C G, TRONCONI E, LIETTI L, et al. Detailed kinetics of the Fischer-Tropsch synthesis on cobalt catalysts based on H-Assisted CO activation[J].Topics in Catalysis, 2011, 54(13): 786-800.
[19] KNOCHEN J, GüTTEL R, KNOBLOCH C, et al. Fischer-Tropsch synthesis in milli-structured fixed-bed reactors: Experimental study and scale-up considerations[J].Chemical Engineering & Processing Process Intensification, 2010, 49(9): 958-964.
[20] YATES I C, SATTERFIELD C N. Intrinsic kinetics of the Fischer-Tropsch synthesis on a cobalt catalyst[J].Energy & Fuels, 1991, 5(1): 168-173.
[21] KAPTEIJN F, DEUGD R M D, MOULIJN J A. Fischer-Tropsch synthesis using monolithic catalysts[J].Catalysis Today, 2005, 105(3-4): 350-356.
[22] MYRSTAD R, ERI S, PFEIFER P, et al. Fischer-Tropsch synthesis in a microstructured reactor[J].Catalysis Today, 2009, 147(9): 301-304.
[23] 錢煒鑫, 張海濤, 應衛勇, 等. 活性炭負載鈷基催化劑費托合成本征動力學[J].天然氣化工(C1化學與化工), 2012, 37(2): 1-6.(QIAN Weixin, ZHANG Haitao, YING Weiyong, et al. Intrinsic kinetics of Fischer-Tropsch synthesis over Co/AC catalyst[J].Natural Gas Chemical Industry, 2012, 37(2): 1-6.)
[24] 馬文平, 劉全生, 趙玉龍, 等. 費托合成反應機理的研究進展[J].內蒙古工業大學學報, 1999, 18(2): 121-127. (MA Wenping, LIU Quansheng, ZHAO Yulong, et al. Recent advances in the study of Fischer-Tropsch synthesis reaction mechanism[J].Journal of Inner Mongolia University of Technology, 1999, 18(2): 121-127.)
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
光學精密工程(2016年6期)2016-11-07 09:07:19
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
核科學與工程(2015年4期)2015-09-26 11:59:03
