擔載Rh量子點三維石墨烯催化劑可見光催化制氫
2018-01-04 21:16:12甄文龍高海波呂功煊馬建泰
無機化學學報 2018年1期
甄文龍 高海波, 呂功煊*, 馬建泰
(1中科院蘭州化學物理研究所,羰基合成與選擇氧化國家重點實驗室,蘭州 730000)
(2蘭州大學化學化工學院,蘭州 730000)
擔載Rh量子點三維石墨烯催化劑可見光催化制氫
甄文龍1高海波1,2呂功煊*,1馬建泰2
(1中科院蘭州化學物理研究所,羰基合成與選擇氧化國家重點實驗室,蘭州 730000)
(2蘭州大學化學化工學院,蘭州 730000)
利用二維石墨烯易彎曲的特性制備了具有拓撲結構的三維石墨烯(3DGR),并將Rh量子點(Rh QDs)負載于其上得到了三維石墨烯Rh量子點光催化劑,用于光催化制氫反應。由于三維石墨烯具有特殊的拓撲結構,消除了電子在石墨烯層中傳遞的各向異性,促進了電子進行層間傳遞,改變了光生電子傳遞路徑,Rh QDs/3DGR催化劑展現出較高的產氫活性和高的穩定性。120 min內累積產氫量為794.2 μmol,在520 nm波長下,表觀量子效率達到12.6%。該催化劑也具有較高的光電流、較低的過電位(-0.34 V)和較長的熒光壽命(1.37 ns)。
三維石墨烯;銠量子點;光催化制氫;拓撲結構
0 引 言
近年來的研究發現,石墨烯具有超高的室溫載流子遷移率,有可能成為替代硅的電子材料[1-5]。石墨烯是碳的一種二維晶體,每個原子通過共價鍵與3個鄰近碳原子連接形成蜂巢晶格,形成富勒烯,納米管和3D石墨等[6]。與許多碳基納米材料一樣,石墨烯展現出許多特殊的電子性質[2-10],可歸因于石墨烯獨特的電子云密度和電子結構,良好的電子快速轉移能力。此外二維石墨烯的拉伸平面結構具有高的強度和剛度,但有非常低的彎曲剛度,這就容易在外部因素的影響下形成裂紋,皺紋和褶皺[11-18]。
石墨烯性質的改變很大程度上也依賴于其表面拓撲結構,不同的拓撲結構會引起熱學、電子傳輸和彈性性能的異常[19]。很多種方法都可以得到拓撲結構的石墨烯[20-21],其中1個是具有莫比烏斯環拓撲結構的石墨烯,它最突出的特征是只有1個邊緣和1個表面,也就是石墨烯的上下表面是等效的[22-24],因而電荷在層間的運移更為有效。三維石墨烯也是具有等效面拓撲結構的碳材料,而關于它作為光催化劑載體的研究還不多見。
太陽能是清潔的可再生能源,光驅動分解水催化制氫被認為是一種新的制氫途徑[25-26],這就要求催化劑具有低的析氫過電位[27-31],快速電子轉移能力[32-35],和對可見光的良好吸收[36]。石墨烯具有快速傳導電子的性能,用于光催化分解水制氫催化劑載體可有效地分離催化劑上產生的電子和空穴,增強電荷的輸運,減少復合的幾率,顯著提升催化劑的產氫性能[37]。
在本文中,我們報道了一種由石墨烯和Rh量子點構成的高效產氫光催化劑,Rh量子點(Rh QDs)是被錨定于具有拓撲結構的三維石墨烯上,由于三維石墨烯(3DGR)具有等效的拓撲結構,明顯地增強了界面間的傳遞電子能力。相比二維石墨烯催化劑(Rh/GR),該催化劑表現出較大的瞬態光電流,較低的析氫過電位(-0.34 V)和較長熒光壽命(1.37 ns),促進了電子在層間傳遞,獲得了優異的光催化產氫性能和較高的產氫表觀量子效率(12.6%)。
1 實驗部分
1.1 試劑和材料
實驗中所用的化學試劑均為分析純,石墨粉,五氧化二磷 (P2O5),硝酸鈉 (NaNO3),高錳酸鉀(KMnO4),水合三氯化銠(RhCl3·xH2O),曙紅 Y 可溶性鈉鹽(EY),過硫酸鉀(K2S2O8),過氧化氫(H2O2),鹽酸(HCl),三乙醇胺(TEOA),無水硫酸鈉(Na2SO4)。 試劑均未進一步純化,直接使用。
1.2 儀器和設備
氫氣在Agilent 6820(TCD)分析,載氣為Ar氣,催化劑的比表面積是在0.05~0.3的相對壓力下利用BET方法測定,孔容是根據相對壓力為0.990的吸附曲線計算得到。采用日本Rigaku B/Max-RB型X射線粉末衍射儀進行測試晶體結構,Cu Kα靶(λ=1.540 6 nm,管電壓為 40 kV,管電流為 40 mA,2θ=10°~80°)。 在美國 ESCALAB 250Xi型 X 射線光電子能譜儀表征催化劑表面化學組成和化學狀態(Mg靶,C1s 284.8 eV校正)。透射電鏡(TEM)和高分辨透射電鏡(HRTEM)表征在Tecnai-G2-F30型場發射透射電鏡上進行(FEI公司,美國),加速電壓為300 kV。FT-IR分析在美國Nexus 870紅外光譜儀上進行,測試波長范圍是4 000到400 cm-1。采用法國Horiba Scientific公司的FluoroMax-4型熒光光譜儀測定穩態熒光光譜。Raman分析在美國Raman 950傅里葉變換激光拉曼光譜儀上進行,最高分辨率:<2 cm-1,中心波長:1 064 nm,波數范圍 100~3 600 cm-1。在吉時利4200 SCS上通過雙探針法測量樣品的電子傳輸性質。載體濃度,遷移率和電阻率通過霍爾效應室溫下用Van der Pauw兩點探針法(Quantum Technology Corp.,Blaine WA,USA)進行測試。
1.3 催化劑的制備
1.3.1 氧化石墨(GO)的制備
采用改進的Hummers法制備氧化石墨(GO)[38-39]。具體過程如下:在攪拌條件下,將20 g石墨粉,10 g K2S2O8和10 g P2O5依次加入到30 mL 80℃的濃硫酸中,反應5 h,待混合物溫度冷卻到室溫后加1 500 mL H2O稀釋,然后抽濾,洗滌直到濾液呈中性,并在室溫下干燥1 d,得到預氧化石墨。在不斷地攪拌及冰浴條件下,將10 g預氧化石墨和5 g NaNO3加入到230 mL預冷至0℃的濃H2SO4中,然后再按照少量多次的辦法加入30 g KMnO4,使得反應溫度不超過20℃。再移去冰水浴,待溫度升高到35℃時持續反應2 h,再加460 mL H2O稀釋,15 min后,再加1 400 mL熱H2O稀釋,同時為了去除未反應的KMnO4,加入25 mL雙氧水(30%,w/w)。最后,過濾混合物,并用 HCl(1∶10,V/V)溶液反復洗滌以除去殘留的金屬離子及酸根離子,然后真空干燥1 d,得到氧化石墨(GO)。
1.3.2 三維石墨烯(3DGR)的制備
如圖 1中(a~b)所示,將 GO 配制成 3 mg·mL-1的分散液,在30 mL的GO分散液中加入120 μL的乙二胺(EDA),超聲分散5 min后,將懸浮液轉移至50 mL水熱反應釜中。加熱至120℃并保持6 h得到三維石墨烯水凝膠。然后通過冷凍干燥,得到三維石墨烯氣凝膠[40]。得到的三維石墨烯經過充分地研磨后待進一步使用。
1.3.3 催化劑Rh/GR和Rh/3DGR的制備

圖1 合成催化劑Rh/3DGR的具體過程示意圖Fig.1 Schematic Illustration of Rh QDs/3DGR photocatalyst synthesis
在3DGR表面擔載Rh納米粒子的具體制備過程如圖1(b~c)所示,在200 mL的石英反應瓶中,加入 100 mL三乙醇胺-水溶液 (10%,V/V,pH=9),再依次加入7 mg GO或3DGR,1 mL RhCl3(Rh含量:2 mg·mL-1)和 70 mg EY,超聲分散 10 min,反應瓶口用硅橡膠密封,反應前用Ar進行鼓泡置換反應瓶除去氧氣40 min,使用300 W的氙燈作為反應光源,并配有420 nm的截止濾光片,在光照原位還原2 h,有效的光照面積大約為10.2 cm2,而后抽濾并用無水乙醇和蒸餾水洗滌,產物在60℃的溫度下真空干燥12 h待用。
1.4 光催化活性測試
光催化分解水制氫反應的活性測試實驗在200 mL的石英反應瓶中進行。加入7 mg GO或3DGR,1 mL RhCl3(Rh 含量:2 mg·mL-1)和 70 mg EY 到 100 mL 三乙醇胺-水(TEOA-H2O)溶液中(10%,V/V,pH=9),超聲分散10 min后,反應瓶用Ar進行鼓泡置換反應瓶內空氣40 min。光催化反應的光源為300 W的氙燈 (HSX-UV300),配備有420 nm的截止濾光片。光催化制氫的反應活性測試是通過一定光照時間內產生的H2的量來計算的,用氣相色譜對氫氣進行分析,檢測器為熱導檢測器(TCD),載氣為Ar氣,分離柱為13X分子篩填充柱,用外標法定量。每隔20 min用進樣針抽取0.5 mL氣體注入GC進行檢測。
測定產氫表觀量子效率AQE時,使用430、460、490、520和550 nm帶通濾光片。并用標定過的FU100型輻射測量儀測定入射光子數,每個濾光片、不同波長下平行測定多次后取平均值。測量光照射到反應瓶上平面窗口的有效面積,然后通過公式(1)計算出一定時間內反應體系所接受的相應光子數,t為光照時間(s),S為反應瓶的有效光照面積(m2),Q為輻射測量儀測定的入射光子數(μmol-1·m-2·s-1)

當計算表觀量子效率時,假定入射的光子全部被體系吸收,并且不做任何的折射散射校正,按照公式(2)計算產氫反應的表觀量子效率(AQE),其中為氫氣的產量(μmol),np為入射光子數。

1.5 光電化學性能測試
線性掃描伏安(LSV)和瞬態光電流測試在CHI 660A電化學工作站進行,實驗采用三電極體系,鉑電極為對電極,飽和甘汞電極(SCE)為參比電極,電解質溶液為 0.1 mol·L-1NaSO4和 10%(V/V)TEOA的混合溶液。將50 μL的5%Nafion和2 mg·mL-1催化劑(EY 濃度為 1.0×10-3mol·L-1)的分散液,經過超聲分散均勻后滴涂在潔凈的導電玻璃(ITO)表面上形成薄層,待樣品干燥后得到工作電極,其有效面積約為0.95 cm2。LSV測量在電勢范圍是-1~0 V進行,掃描速率為1 mV·s-1。瞬態光電流測試光源為300 W氙燈且配有420 nm的截止濾光片。
2 結果與討論
2.1GO與3DGR結構與性能對比
催化劑拉曼光譜如圖2(a)所示,位于1 350和1 594 cm-1峰可歸屬為石墨烯的D峰和G峰,分別反映sp3碳原子和sp2雜化石墨碳原子表面的缺陷程度[41-43]。3DGR的拉曼光譜在1 322和1 595 cm-1處有兩個特征峰,D峰發生位移反映了由于C-C和C-N鍵距離改變引起的石墨烯片結構變形。從Tuinstra-Koenig(TK)方程得知,D峰與G峰的強度比(ID/IG)可用于估算石墨烯的無序化程度[37]。3DGR的ID/IG(1.23)要大于GR的ID/IG(0.90),這表明與GO相比,由于EDA的還原引起了3DGR的缺陷,無序度增加。為了證明通過EDA水熱還原形成了三維石墨烯,作者測試了樣品的傅里葉變換紅外光譜(FTIR),結果如圖2(b)所示。在氧化石墨樣品中觀察到OH伸縮振動峰(3 500~3 000 cm-1),CH伸縮振動峰(2 980~2 900 cm-1),C=O從羰基和羧基伸縮振動峰(~1 730 cm-1),芳族 C=C 伸縮振動峰(~1 619 cm-1),羧基 CO(~1 390 cm-1)峰,環氧基(~1 258 cm-1)和烷氧基(~1 062 cm-1)的CO伸縮振動峰[43-46]。位于2 358 cm-1處的強吸收峰可能來自二氧化碳中C=O雙鍵的伸縮振動[47]。通過EDA還原后,含氧基團的所有特征吸收帶強度均顯著降低,并且出現位于1 559 cm-1處的石墨烯骨架振動的特征峰,表明GO已被成功地還原為3DGR。圖2(c)顯示了GO和3DGR的C1s XPS峰,表明相關碳為石墨烯。3DGR的XPS譜表明,3DGR中一部分C-O-C基團(286.4 eV)轉化為C-N基團(285.6 eV),由于氮的加入使得3DGR電子結構無序性增加。C-N的存在是因為GO表面含有少量的羧基,經過乙二胺還原成,可能會形成酰胺鍵。XPS的C1s譜進一步證實了富氧基團的存在,如 C-O-C(3DGR 為 286.4 eV,GO 為 286.8 eV),C=O(287.7 eV)和 O-C=O(288.8 eV)[48-49]。 此外,根據 3DGR和GO的C=C峰可以得知,3DGR中C=C官能團的相對含量增加,表明離域的范圍增加,提高了材料平面的電導率[36]。GO和3DGR電極的奈奎斯特圖(Nyquist plots)如圖2(d)所示。與GO電極相比,3DGR在高到中頻區域內顯示了最小的半圓,表明三維框架結構進一步促進了石墨烯層間的電荷轉移。

圖2 GO和3DGR的Raman(a),FTIR(b),C1s XPS(c)和EIS(d)Fig.2 Raman(a),FTIR(b),C1s XPS(c)and EIS(d)spectra of GO and 3DGR samples

表1 樣品的結構表征分析Table1 Structural analysis of GO and 3DGR
表1給出了所有樣品的比表面積(SBET)和孔體積等數據,從表中可以發現,GO、3DGR和Rh QDs/3DGR催化劑的比表面積分別為132、220和186 m2·g-1。 此外,3DGR 存在較大的孔體積(0.180 cm3·g-1)和較小的孔尺寸 (3.28 nm)。與3DGR相比,Rh QDs/3DGR催化劑的比表面積、孔體積、孔尺寸均略有減小,可能是Rh進入3DGR的三維孔道內部所致,進而為光催化分解水產氫提供了更多的活性位點,有利于光催化性能的提升。
圖3是77 K下GO和3DGR的N2吸附-脫附等溫曲線和孔徑分布圖。從圖3(a)中可以看出,兩種樣品的吸附-脫附等溫線均呈現Ⅳ型,H3型滯后環,說明制備的樣品中存在介孔結構。從圖3(b)中看出,GO的孔徑主要是2.1和3.2 nm,而通過乙二胺還原成三維石墨烯之后,孔徑除了2.1 nm之外,還有4.6、10.7和20.1 nm,表明3DGR具有較大的孔隙。

圖3 77 K下催化劑GO或3DGR的N2吸附-脫附等溫線(a)和孔徑分布圖(b)Fig.3 (a)N2adsorption-desorption isotherms;(b)Pore size distributions of GO and 3DGR catalysts measured at 77 K

圖4 在可見光照射(λ≥420 nm)下,在100 mL(10%,V/V,pH 9)的TEOA溶液中,助催化劑EY-Rh,EY-Rh/GR和EY-Rh/3DGR的產氫活性圖Fig.4 Time courses of hydrogen evolution over EY-Rh,EY-Rh/GR and EY-Rh/3DGR photocatalysts in 100 mL 10%(V/V)TEOA-H2O solution(pH=9)under visible light irradiation(λ≥420 nm)
2.2 光催化產氫的反應活性和不同pH對反應速率的影響
光催化產氫反應活性測試是在以TEOA為犧牲試劑,EY為光敏化劑,可見光(λ≥420 nm)照射條件下完成的。圖4是所有樣品產氫總量與反應時間關系圖,從圖中可以發現Rh與Rh/GR催化劑在120 min內的產氫量分別為98.9和171.2 μmol。而Rh/3DGR催化劑具有最高的產氫效率,在120 min內產氫總量為794.2 μmol。對于相同量Rh的催化劑,催化劑EY-Rh/3DGR的產氫活性是EY-Rh活性的8倍,是EY-Rh/GR的近5倍。這表明高效產氫催化劑Rh QDs/3DGR,3DGR的拓撲結構促進了電子的快速傳遞,進而明顯增強光催化分解水產氫的速率。另外3DGR存在大共軛π鍵,它與EY分子之間的相互作用可促進電子通過拓撲結構快速轉移至Rh表面[50-51]。
為了探究Rh擔載量對光催化反應活性的影響,我們制備了不同Rh擔載量的Rh/3DGR催化劑。不同Rh負載量下催化劑的產氫活性如圖5所示,當Rh的擔載量為2 mg時,單位質量Rh的表面最大光催化產氫速率是 198.6 mmol·h-1·g-1。 當 Rh含量為4或6 mg時,產氫速率有明顯地下降,可能是因為部分Rh組分發生團聚,所以擔載量進一步增大時活性反而降低。
在光解水反應體系中,溶液pH可以明顯影響光催化產氫反應的活性[52]。在圖6中,當TEOA的pH值從5到13變化時,產氫速率在pH=9時達到最大(2 h內產氫量為794.2 μmol),在偏酸或偏堿條件下產氫速率都會減小。酸性條件下,產氫活性低主要是因為TEOA的質子化而降低了激發態EY*的熒光壽命。在堿性條件下,水溶液中由于存在大量OH-使得H+減少,使得產氫速率降低[45]。此外,pH可能還會影響EY在催化劑表面的吸附,Min等[51-53]報道了EY的吸附主要是依賴于EY和載體之間的π-π堆積的非共價鍵相互作用。酸性條件下,3DGR拓撲結構上存在官能團(如羧基)易被質子化;強堿性條件下,EY分子表面存在的羧基會失去質子以及3DGR表面帶有負電荷阻礙H+的吸附[54-55]。
2.3 表觀量子效率(AQEs)和穩定性測試
為了研究催化劑在不同波長輻照時的產氫速率,作者比較了Rh/3DGR和Rh/GR在不同波長下(430,460,490,520 和 550 nm) 的 表 觀 量 子 效 率(AQEs)。結果如圖7所示,Rh/3DGR在每個波長下的AQE均高于Rh/GR。在430 nm波長下,催化劑Rh/3DGR和Rh/GR的AQE分別為9.9%和2.8%,在520 nm處,催化劑Rh/3DGR和Rh/GR展現出最高的AQE,分別為12.6%和4.3%,這主要是因為EY的最大吸收波長在520 nm附近[56]。
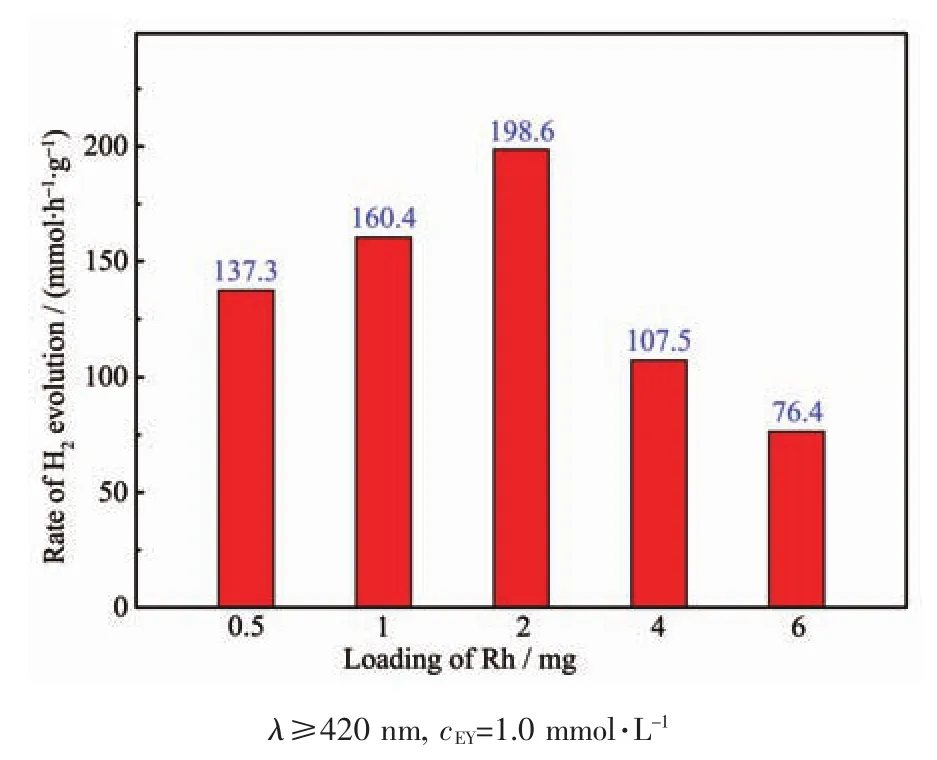
圖5 在可見光條件下,EY敏化不同Rh擔載量助催化劑Rh/3DGR在100 mL 10%(V/V)的TEOA水溶液(pH=9)中2 h內的產氫活性圖Fig.5 Effect of the loading of Rh on photocatalytic rate of H2evolution over EY-Rh/3DGR photocatalysts in 100 mL 10%(V/V)TEOA aqueous solution(pH 9)under visible light irradiation

圖6 在可見光條件下,100 mL(10%,V/V)的TEOA溶液的不同pH對EY敏化Rh/3DGR光催化產氫速率的影響Fig.6 Effect of pH value on photocatalytic rate of H2 evolution over EY-Rh/3DGR photocatalyst in 100 mL 10%(V/V)TEOA aqueous solution under visible light irradiation

圖7 不同波長下,在100 mL(10%,V/V)的TEOA溶液中,EY(1.0 mmol·L-1)敏化 Rh/3DGR 和Rh/GR體系產氫量子效率Fig.7 AQE of hydrogen evolution for EY(1.0×10-3mol·L-1)sensitized systems by Rh/3DGR,Rh/GR,in 100 mL of 10%TEOA-H2O solution under different wavelength irradiation
催化劑Rh/3DGR和Rh/GR的穩定性測試結果如圖8所示。在第一輪循環(120 min內),Rh/3DGR產氫總量達到了794.2 μmol,而此時Rh/GR的產氫量只有171.2 μmol。而后對反應溶液進行抽濾并用無水乙醇和蒸餾水多次洗滌催化劑,然后加入新的EY和TEOA溶液進行超聲分散后,用Ar氣置換反應瓶內氣體。測試第二輪循環,催化劑Rh/3DGR和Rh/GR的產氫總量分別為802.2和192.2 μmol。這可能是由于在第一輪循環中,Rh的還原消耗了少量的H2。進行第三和第四輪循環,可能是在抽濾和洗滌過程中催化劑的損失造成了產氫活性逐漸下降。因此,催化劑Rh/3DGR在光催化析氫反應中依然保持了較好的穩定性。

圖8 可見光下(λ≥420 nm),在100 mL 10%(V/V,pH 9)TEOA溶液中,測試了催化劑Rh/3DGR(紅色小球)和Rh/GR(藍色小球)產氫的穩定性Fig.8 Stability tests of hydrogen evolution over EY photosensitized Rh/3DGR(red ball)and Rh/GR(blue ball)under visible light irradiation(λ≥420 nm)
2.4 催化劑的電導率和霍爾效應分析
為了研究3DGR拓撲結構對導電性的影響,通過雙探針法進行測試了樣品的電子傳輸性能。(如圖9所示)。在空氣中,室溫下在玻璃基底上的金接觸點處設置圓柱體。明顯地,與原始GO相比,3DGR的電流增加了3個數量級,表明經過水熱還原處理明顯改變了石墨烯的電性質。負載Rh之后,催化劑的電流增大了3個數量級,表明增強了電子傳遞。此外,當掃描電壓較小(約-1~0 V)時,GO的電流具有劇烈的變化和波動。掃描電壓增加時,GO具有低電流、I-V曲線趨于平滑和穩定。GO的I-V特性曲線整個范圍內不對稱,表現出肖特基勢壘特性,也反映出樣品的半導體特性[57],高動能的電子可以在更高的掃描電壓下穿過原始GO的肖特基勢壘。對于較低的掃描電壓,會發生相反的現象。催化劑3DGR、Rh/GR和Rh/3DGR的I-V曲線相對平滑且對稱,表明樣品的電阻相對較小。也就是說催化劑3DGR、Rh/GR和Rh/3DGR表現出歐姆特性。通過水熱還原GO形成具有拓撲結構的3DGR改善了導電性和遷移率。
為了進一步研究樣品的電學性能,測試了霍爾效應。我們發現3DGR、Rh/GR和Rh/3DGR顯示出p型導電性能,而GO幾乎是絕緣材料。Rh/GR和Rh/3DGR的載流子濃度分別為 3.609×1017和1.478×1019cm-3, 遷移率分別為 20.38和 0.511 2 cm2·V-1·s-1。在之前的研究中,發現石墨烯的電性能可以通過還原反應逐漸增強[58-59]。然而3DGR通過水熱法乙二胺還原GO,對石墨烯二維片層結構進行了重組,構建三維結構,很大程度上提高了3DGR電子傳輸性能,載流子濃度,導電性和遷移率。

圖9 催化劑GO,3DGR,Rh/GR和Rh/3DGR的I-V特性Fig.9 I-V curve of GO,3DGR,Rh/GR and Rh/3DGR catalysts

圖10 樣品GO,3DGR,Rh/GR和Rh/3DGR的XRD圖Fig.10 XRD patterns of GO,3DGR,Rh/GR and Rh/3DGR
2.5 催化劑的XRD和XPS分析
我們測試了反應前后樣品的X射線粉末衍射(XRD),結果如圖10所示。從GO的XRD結果中可以看出,2θ在12.5°附近有較強的衍射峰,屬于GO的特征衍射峰,這與相關文獻中報道的XRD結果一致[60]。催化劑3DGR、Rh/GR和Rh/3DGR的衍射峰主要位于23.67°,與相關還原氧化石墨烯的衍射峰較為接近,說明GO被還原為具有拓撲結構的三維石墨烯[61]。此外,對XRD衍射峰圖像放大,還可以發現位于40.87°的衍射峰,可被歸屬為面心立方結構的 Rh(111)晶面(PDF#88-2334)。在催化劑 Rh/3DGR中,由于Rh納米粒子尺寸為量子尺寸,因此導致衍射峰變弱,以至于沒有觀測到。而在Rh/GR中,觀測到Rh的衍射峰,說明Rh被還原在GR表面時形成的顆粒比在3DGR表面的顆粒尺寸要大。
為了進一步探究已制備的催化劑Rh/3DGR的Rh化學狀態,測試X射線光電子能譜(XPS),結果如圖11所示,催化劑Rh/3DGR的Rh3d精細譜結合能位于307.3和312.1 eV,分別歸屬于Rh3d5/2和Rh3d3/2[62],表明在3DGR表面Rh是以金屬態的形式存在。

圖11 催化劑Rh/3DGR的Rh3d精細XPS能譜Fig.11 Rh3d XPS spectrum for Rh/3DGR photocatalyst

圖12 催化劑GO或Rh/GR的掃描電鏡形貌Fig.12 SEM images of GO and Rh/GR
2.6 催化劑的SEM,TEM,元素Mapping和EDX分析
圖12是GO和Rh/GR的SEM圖,從圖12(a)可以看出,GO為有許多凸凹不平的層狀褶皺,為多層重疊,具有一定的微觀粗糙度。但片層表面密實,少有孔狀結構,經過負載Rh之后,如圖12(b)所示,石墨烯表面出現了較多相對較小、均勻顆粒。這可能使納米粒子的沉積與團聚造成。
圖13展示了催化劑Rh/3DGR的高分辨透射電子顯微鏡(HRTEM)圖像和能量色散X射線(EDX)結果。通過圖13(a)可以發現,3DGR具有三維結構,石墨烯片進行空間折疊、旋轉或卷曲,表現出許多孔狀且比較蓬松的結構,其周邊同樣有許多凸凹不平的層狀褶皺的拓撲結構。通過圖13(b~d)可以發現,Rh納米顆粒均勻分布在3DGR的表面,從HRTEM照片中可以看到Rh相鄰兩晶面的間距為0.223 nm,可歸屬為Rh(111)晶面[63]。通過粒徑分布統計可以得到Rh納米粒子的平均尺寸約為1.2 nm,證明其為Rh QDs。從高角環形暗場-掃描透射電子顯微鏡 (HAADF-STEM)圖像和EDX元素Mapping圖中可以發現元素C、N、O、Cu和Rh在催化劑Rh/3DGR中的分布是非常均勻的(圖13(e))。另外圖13(f)的EDX結果也證實了在催化劑Rh/3DGR中存在Rh元素。
2.7 瞬態光電流-時間響應和線性掃描伏安法
為了進一步研究電子轉移機制,我們將催化劑Rh/GR和Rh/3DGR涂在導電玻璃ITO上制成工作電極,在可見光照射下,測試了EY敏化下的瞬態光電流-時間響應曲線(如圖14)。ITO/EY-Rh/GR電極比ITO電極展示出顯著的光電流響應,而ITO/EYRh/3DGR電極展示出了最高的光電流。結果表明,高的H2產量可歸因于Rh QDs的高分散以及3DGR的拓撲結構增強了電子的傳遞,使得界面電子快速的從EY-傳遞到Rh QDs進而生成H2。正如在圖9中討論的一樣,3DGR在形成三維結構后可以明顯的改善電子傳遞速率,降低材料的電阻,進而優化Rh QDs表面催化活性位點產生優異的光電流。
此外,我們還用線性掃描伏安法(LSV)研究了ITO,Rh/GR和Rh/3DGR電極的電催化產氫活性,結果如圖15所示。隨著電壓的降低,單獨的ITO電極將水還原成H2產生的陰極電流數值很低。電極ITO-Rh/GR顯示,在相近電勢范圍內,陰極電流有明顯增加,其析氫過電位為-0.40 V,表明催化劑Rh/GR是有利于電子轉移且可以達到高效分解水產氫的目的。電極ITO-Rh/3DGR展現出最低的析氫過電位(-0.34 V),暗示著Rh/3DGR催化劑在光催化分解水制氫反應中有更高的活性。在高度分散的活性組分Rh QDs表面形成氫氣的過程中,Rh/3DGR擁有的低過電位可能是由于Rh QDs和3DGR拓撲結構之間的快速電子傳遞以及更加穩定利于H+快速吸附的活性中間體,使得H2O/H+快速還原成H2。這一結果與光電流測試結果相一致,意味著Rh/3DGR助催化劑能夠快速地傳遞電子并使之發生水的還原反應。

圖13 3DGR的SEM(a),催化劑Rh/3DGR的TEM(b),HRTEM(c),Rh的粒徑分布(d),HAADF-STEM和元素 mapping(e)和 EDX(f)Fig.13 SEM images of 3DGR(a),TEM(b),HRTEM images(c),particle size distribution of Rh(d),HAADF-STEM and elemental mapping(e)and EDX spectrum(f)of Rh/3DGR samples

圖14 工作電極EY-Rh/GR和EY-Rh/3DGR在10%(V/V,pH 9)TEOA 和 Na2SO4(0.1 mol·L-1)混合電解質溶液中,在可見光照射下測定的光電流-時間曲線Fig.14 Transient photocurrent-time profiles of EY-sensitized Rh/GR and Rh/3DGR in a mixed solution of 10%(V/V)TEOA and Na2SO4(0.1 mol·L-1)at pH=9 under visible light irradiation(λ≥420 nm)

圖15 在 10%(V/V,pH=9)TEOA 和 Na2SO4(0.1 mol·L-1)混合電解質溶液中工作電極ITO,Rh/GR和Rh/3DGR的線性掃描伏安曲線Fig.15 LSV curves of bare ITO glass and Rh/GR and Rh/3DGR coated on ITO electrodes in a mixed solution of 10%(V/V)TEOA and 0.1 mol·L-1 Na2SO4at pH=9

圖16 EY敏化Rh/GR和Rh/3DGR體系的穩態熒光光譜Fig.16 Photoluminescence quenching of EY by Rh/GR and Rh/3DGR in 10%(V/V)TEOA-H2O solution at pH=9
2.8 穩態熒光和瞬態熒光光譜分析
為了研究3DGR在光生電子轉移中的作用,測試了EY敏化助催化劑Rh/GR和Rh/3DGR的穩態熒光(PL)光譜(如圖16)。當激發波長為480 nm時,EY水溶液在534 nm處顯示出最強的發射峰,對應EY激發態電子的回躍淬滅過程。EY吸收能量后,電子從基態躍遷至激發態,由于EY激發態電子壽命很短,若沒有被及時而有效的轉移時,就會以輻射的形式釋放能量而使得熒光發生淬滅[64-65]。當向EY中加入催化劑Rh/GR和Rh/3DGR后,EY的發射峰強度有顯著降低,其中EY+Rh/GR的強度最高,而Rh/3DGR對應的峰強度是最低的。結果表明在Rh/3DGR上激發電荷的復合比其它催化劑上要慢,這是由于3DGR與EY之間形成強的非共價ππ堆積相互作用。通過這種作用,電子從EY*轉移到3DGR拓撲結構和Rh QDs的表面。因此,電子通過激發態染料與3DGR之間的π-π共軛相互作用被快速傳遞至3DGR拓撲結構上,最終電子被快速轉移至Rh QDs表面還原水產氫。
采用瞬態熒光光譜技術對催化劑Rh/GR和Rh/3DGR與EY之間的相互作用進行了研究分析,表2是催化劑的熒光壽命數據。除了單獨的EY溶液采用一級指數擬合,其余添加了催化劑的EY溶液均采用二級指數擬合。在表2中,單重激發態的EY1*壽命是1.18 ns,其它催化劑的熒光壽命有所增加,依次是 1.20 ns(GR)、1.25 ns(3DGR)和 1.28 ns(Rh/GR)。然而當向EY溶液內加入Rh/3DGR,EY的熒光壽命有了明顯的增加 (1.37 ns)。催化劑Rh/3DGR的短壽命(1.20 ns)和長壽命(1.52 ns)可能來源于已結合3DGR的EY和未結合3DGR的EY,擬合結果有2個指數衰減函數表示動態熒光猝滅。Lazarides等研究發現在加入TEOA或催化劑后,單線態的熒光染料不會發生淬滅,這也進一步說明了EY1*不涉及電子轉移[66],最初形成的EY1*經過系間竄越過程變為三重激發態 (EY3*)。這些結果表明,3DGR可以明顯延長Rh/3DGR系統中EY1*的壽命,很大程度上促進了EY1*系間竄越產生EY3*,然后得到電子形成EY-。憑借著3DGR快速的電子傳遞能力,電子從EY-傳遞到3DGR和活性組分(Rh QDs)表面,使得質子被還原為H2分子。

表2GO,3DGR,Rh/GR和Rh/3DGR樣品在EY的TEOA水溶液中的熒光壽命Table2 Decay parameters of EY in the presence of GO,3DGR,Rh/GR and Rh/3DGR samples
2.9 催化劑Rh/3DGR的光催化產氫機理
圖17給出了催化劑Rh/3DGR在EY敏化下的可見光催化制氫的可能機理。3DGR具有較大的比表面積~220 m2·g-1,利于電子進行層間傳遞的拓撲結構和高的電導率等特性,極大地提高了電子傳遞效率。在可見光照射下,EY吸收能量后形成單重激發態的EY1*,經過系間竄躍(ISC)過程形成 EY3*,在TEOA 的 存 在 下 ,EY3*得 到 電 子 形 成 EY·-[66-68]。3DGR具有快速傳遞電子的特性,優先將EY·-中的電子轉移至3DGR。在3DGR框架累積的電子被快速傳遞到活性組分Rh QDs表面,最終水中的質子得到來自Rh QDs的電子被還原成H2。3DGR作為一個優秀的電子受體和轉運體可以有效地延長電荷載體的壽命并促進電荷有效分離,貴金屬量子點Rh QDs具有低的氫吸附自由能,最終使得Rh/3DGR展現出了高的產氫活性與穩定性。

圖17 Rh/3DGR在EY敏化體系中可見光條件下高效制氫的機理Fig.17 Mechanism of HER over EY sensitized Rh/3DGR under visible illumination
3 結 論
在本文中,我們通過水熱法利用二維石墨烯的缺陷和褶皺合成了具有拓撲結構的三維石墨烯(3DGR),將高效助催化劑Rh量子點(Rh QDs)錨定在3DGR上用來光催化分解水產氫。相比二維石墨烯催化劑Rh/GR,催化劑Rh/3DGR表現出較大的瞬態光電流,較低的產氫過電位(-0.34 V)和較長時間熒光壽命(1.37 ns)。3DGR明顯地增強了材料傳遞電子的能力,具有拓撲結構的3D石墨烯可以增強電子通過層間傳遞,進而促進光生電子快速的從激發態EY(EY*)傳遞到Rh QDs。催化劑Rh/3DGR獲得了優異的光催化產氫性能 (2 h內積累產氫量為794.2 μmol)和在520 nm的光照射下的最高的表觀量子效率(~12.6%)。
[1]Novoselov K S,Geim A K,Morozov S V,et al.Science,2004,306(5696):666-669
[2]Zhang Y,Tan Y W,Stormer H L,et al.Nature,2005,438(7065):201-204
[3]Chen J H,Jang C,Xiao S,et al.Nat.Nanotechnol.,2008,3(4):206-209
[4]Ni Z H,Yu T,Lu Y H,et al.ACS Nano,2008,2(11):2301-2305
[5]Quhe R,Zheng J,Luo G,et al.NPG Asia Mater.,2012,4(2):e6
[6]Geim A K,Novoselov K S.Nat.Mater.,2007,6(3):183-191
[7]Novoselov K S,Geim A K,Morozov S V,et al.Nature,2005,438(7065):197-200
[8]Neto A H C,Guinea F,Peres N M R,et al.Rev.Mod.Phys.,2009,81(1):109
[9]Weller T E,Ellerby M,Saxena S S,et al.Nat.Phys.,2005,1(1):39-41
[10]Katsnelson M I,Novoselov K S,Geim A K.Nat.Phys.,2006,2(9):620-625
[11]Partovi-Azar P,Nafari N,Tabar M R R.Phys.Rev.B,2011,83(16):165434
[12]WANG Qing-Yun(王清云),TONG Yong-Chun(佟永純),XU Xin-Jian(徐新建),et al.J.Mol.Catal.(China)(分子催化),2016,30(1):80-87
[13]Costamagna S,Neek-Amal M,Los J H,et al.Phys.Rev.B,2012,86(4):041408
[14]YANG Qin(楊琴),ZHOU Juan(周娟),YIN Meng-Yun(尹夢云),et al.J.Mol Catal.(China)(分子催化),2016,30(2):99-104
[15]Baimova J A,Dmitriev S V,Zhou K,et al.Phys.Rev.B,2012,86(3):035427
[16]NI Jun(倪軍),LUO Xiao-Fang(羅曉小),ZHAN Yong(詹勇),et al.J.Mol Catal.(China)(分子催化),2016,30(3):282-296
[17]Baimova J A,Liu B,Dmitriev S V,et al.Phys.Status Solidi RRL,2014,8(4):336-340
[18]Lü Gong-Xuan(呂功煊),TIAN Bin(田彬).J.Mol.Catal.(China)(分子催化),2017,31(2):101-104
[19]Blanter Y M,Kaganov M I,Pantsulaya A V,et al.Phys.Rep.,1994,245(4):159-257
[20]Tombros N,Jozsa C,Popinciuc M,et al.Nature,2007,448(7153):571-574
[21]Trauzettel B,Bulaev D V,Loss D,et al.Nat.Phys.,2007,3(3):192-196
[22]Ajami D,Oeckler O,Simon A,et al.Nature,2003,426(6968):819-821
[23]Starostin E L,Van Der Heijden G H M.Nat.Mater.,2007,6(8):563-567
[24]Guo Z L,Gong Z R,Dong H,et al.Phys.Rev.B,2009,80(19):195310
[25]Esswein A J,Nocera D G.Chem.Rev.,2007,107(10):4022-4047
[26]Lü Gong-Xuan(呂功煊),ZHANG Wen-Yan(張文妍).J.Mol.Catal.(China)(分子催化),2017,31(5):401-410
[27]Zhang L,Wu H B,Yan Y,et al.Energy Environ.Sci.,2014,7(10):3302-3306
[28]HE Ping(何平),CHEN Yong(陳勇),FU Wen-Fu(傅文甫).J.Mol.Catal.(China)(分子催化),2016,30(3):269-275
[29]LU Qiang(盧強),LI Cao-Long(李曹龍),WANG Fei(王飛),et al.J.Mol.Catal.(China)(分子催化),2016,30(6):557-565
[30]Zhen W,Ma J,Lu G.Appl.Catal.B,2016,190:12-25
[31]Tian B,Li Z,Zhen W,et al.J.Phys.Chem.C,2016,120(12):6409-6415
[32]Zhen W,Gao H,Tian B,et al.ACS Appl.Mater.Interfaces,2016,8(17):10808-10819
[33]Li Z,Tian B,Zhang W,et al.Appl.Catal.B,2017,204:33-42
[34]Kong C,Min S,Lu G.ACS Catal.,2014,4(8):2763-2769
[35]Min S,Lu G.Int.J.Hydrogen Energy,2013,38(5):2106-2116
[36]Niu P,Yin L C,Yang Y Q,et al.Adv.Mater.,2014,26(47):8046-8052
[37]Kong C,Min S,Lu G.Chem.Commun.,2014,50(39):5037-5039
[38]Min S,Lu G.Int.J.Hydrogen Energy,2012,37(14):10564-10574
[39]Kong C,Li Z,Lu G.Int.J.Hydrogen Energy,2015,40(31):9634-9641
[40]Zhu X,Zhang P,Xu S,et al.ACS Appl.Mater.Interfaces,2014,6(14):11665-11674
[41]Cai D,Wang S,Lian P,et al.Electrochim.Acta,2013,90:492-497
[42]Wang H,Zhang C,Liu Z,et al.J.Mater.Chem.,2011,21(14):5430-5434
[43]Yen M Y,Hsieh C K,Teng C C,et al.RSC Adv.,2012,2(7):2725-2728
[44]Wang X,Weng Q,Liu X,et al.Nano Lett.,2014,14(3):1164-1171
[45]Park S H,Bak S M,Kim K H,et al.J.Mater.Chem.,2011,21(3):680-686
[46]Wu X,Zhou J,Xing W,et al.J.Mater.Chem.,2012,22(43):23186-23193
[47]Gao H,Zhen W,Ma J,et al.Appl.Catal.B,2017,206:353-363
[48]Chen S,Duan J,Ran J,et al.Energy Environ.Sci.,2013,6(12):3693-3699
[49]Li Y,Zhao Y,Cheng H,et al.J.Am.Chem.Soc.,2011,134(1):15-18
[50]Hirai K,Falcaro P,Kitagawa S,et al.Struct.Bond.,2014,157:167-186
[51]Kataoka Y,Sato K,Miyazaki Y,et al.Energy Environ.Sci.,2009,2(4):397-400
[52]Min S,Lu G.J.Phys.Chem.C,2011,115(28):13938-13945
[53]Zhang H,Lv X,Li Y,et al.ACS Nano,2009,4(1):380-386
[54]Li Q,Chen L,Lu G.J.Phys.Chem.C,2007,111(30):11494-11499
[55]Li D,Müller M B,Gilje S,et al.Nat.Nanotechnol.,2008,3(2):101-105
[56]Li Z,Tian B,Zhen W,et al.Appl.Catal.B,2017,203:408-415
[57]Zhang X,Lu G.Carbon,2016,108:215-224
[58]Kong C,Li Z,Lu G.Int.J.Hydrogen Energy,2015,40(17):5824-5830
[59]Li Z,Wang Q,Kong C,et al.J.Phys.Chem.C,2015,119(24):13561-13568
[60]Yeh T F,Syu J M,Cheng C,et al.Adv.Funct.Mater.,2010,20(14):2255-2262
[61]Liu S,Zeng T H,Hofmann M,et al.ACS Nano,2011,5(9):6971-6980
[62]Strohm J J,Zheng J,Song C.J.Catal.,2006,238(2):309-320
[63]Zhang Y,Grass M E,Habas S E,et al.J.Phys.Chem.C,2007,111(33):12243-12253
[64]Li Z,Wu Y,Lu G.Appl.Catal.B,2016,188:56-64
[65]Tian B,Zhen W,Gao H,et al.Appl.Catal.B,2017,203:789-797
[66]Lazarides T,McCormick T,Du P,et al.J.Am.Chem.Soc.,2009,131(26):9192-9194
[67]Zhang W,Hong J,Zheng J,et al.J.Am.Chem.Soc.,2011,133(51):20680-20683
[68]Wang D Y,Gong M,Chou H L,et al.J.Am.Chem.Soc.,2015,137(4):1587-1592
Photocatalytic Hydrogen Evolution over Rh Quantum Dots Loaded on 3D Graphene under Visible Light Irradiation
ZHEN Wen-Long1GAO Hai-Bo1,2Lü Gong-Xuan*,1MA Jian-Tai
(1State Key Laboratory for Oxo Synthesis and Selective Oxidation,Lanzhou Institute of Chemical Physics,Chinese Academy of Science,Lanzhou 730000,China)
(2College of Chemistry and Chemical Engineering,Lanzhou University,Lanzhou 730000,China)
Based on the two-dimensional graphene′s flexible properties,we constructed the 3D graphene with topological structure,and loaded the Rh QDs on it to prepare the Rh QDs/3DGR catalyst,which was used for the photocatalytic hydrogen production reaction.The topological structure of 3D graphene could eliminate anisotropy of electrons and promote the electrons transfer in the graphene layers,which improved the electron transfer rate and the activity and stability of catalyst.The amount of H2reached 794.2 μmol in 120 min over EY-sensitized Rh/3DGR catalyst,and the corresponding AQE was 12.6% (520 nm).In addition,the Rh/3DGR catalyst showed the largest transient photocurrent,the lowest overpotential(-0.34 V)and the longest fluorescence lifetime(1.37 ns).
3D graphene;Rh QDs;photocatalytic hydrogen generation;topological structure
O614.82+2
A
1001-4861(2018)01-0020-13
10.11862/CJIC.2018.024
2017-08-25。收修改稿日期:2017-11-20。
國家自然科學基金(No.21673262,21173262)和國家自然科學基金重點項目(No.21433007)資助。
*通信聯系人。 E-mail:gxlu@lzb.ac.cn,Tel./fax:+86-931-4968178
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
