樺木酸納米粒的制備與表征
2017-11-16 06:36:13吳銘芳許文佳祖元剛趙修華東北林業大學森林植物生態學教育部重點實驗室哈爾濱150040
中國藥房 2017年31期
吳銘芳,許文佳,祖元剛,趙修華(東北林業大學森林植物生態學教育部重點實驗室,哈爾濱150040)
樺木酸納米粒的制備與表征
吳銘芳*,許文佳,祖元剛,趙修華#(東北林業大學森林植物生態學教育部重點實驗室,哈爾濱150040)
目的:制備樺木酸納米粒,并對其進行表征。方法:以乙醇為溶劑、水為反溶劑,采用反溶劑重結晶法制備樺木酸納米粒。以粒徑為指標,采用單因素試驗與正交試驗優化樺木酸納米粒處方工藝中樺木酸溶液質量濃度、反溶劑-溶劑體積比、反溶劑滴加速度、反應溫度和攪拌速度,并進行驗證試驗。采用掃描電子顯微鏡、激光粒度儀、傅里葉紅外光譜儀和質譜分析儀對所制得的樺木酸納米粒進行表征。結果:最優處方工藝為樺木酸溶液質量濃度為3 mg/mL、反溶劑-溶劑體積比為1∶1、反溶劑滴加速度為8 mL/min、反應溫度為20℃、攪拌速度為900 r/min;所制樺木酸納米混懸液粒徑為(156.0±8.6)nm(n=3),凍干后粒徑為(235.0±12.2)nm(n=3),外觀近球形、大小均勻、形態較規整;與樺木酸原料藥比較,所制樺木酸納米粒的化學結構沒有發生改變,分子量和質荷比無明顯變化。結論:成功制得樺木酸納米粒。
樺木酸;納米粒;反溶劑重結晶法;單因素試驗;正交試驗;表征
樺木酸(Betulinic acid)又稱白樺酯酸,是從白樺樹皮中萃取得到的一種五環三萜類化合物,具有抗腫瘤[1]、抗人類免疫缺陷病毒[2]、抗炎[3-4]和抗瘧病[5]等藥理活性。研究發現,樺木酸能夠抑制腫瘤細胞生長和對抗血管新生,從而誘導癌細胞凋亡,對癌細胞具有靶向作用,且毒性很小,是最有前途的抗癌藥物前體之一。但由于樺木酸水溶性差、口服生物利用度低[6],其應用受到了限制。為了提高難溶性藥物的生物利用度,可將其制備成粒徑小、粒度分布均勻的納米粒。藥物納米化的常用方法有氣流粉碎法、機械粉碎法、高壓均質技術、超臨界流體技術和反溶劑重結晶法等。目前應用最廣泛的是機械粉碎法,但是其缺點是耗能大、效率低和所制納米粒粒徑分布不均勻;而高壓均質技術[7]和超臨界流體技術[8]又都存在高投資、低產率等缺點。
反溶劑重結晶法是把藥物溶于溶劑中后,再加入反溶劑中,通過降低溶劑的溶解能力,使藥物迅速達到過飽和狀態而析出結晶的一種傳統微粉化技術[9]。此方法具有易操作、成本低及能夠適應工業化大生產等優點,通過該法已成功制備了多種難溶性口服藥物納米粒[10]。本研究擬通過反溶劑重結晶法制備樺木酸納米粒并對其進行表征,以期提高樺木酸的口服生物利用度。
1 材料
1.1 儀器
BI-200SM型Zeta電位及粒度分析儀(美國Brookhaven公司);78HW-1型數顯磁力攪拌器(杭州儀表電機有限公司);BS-110型電子分析天平(德國Sartorius公司);Inspect S型掃描電子顯微鏡(SEM,美國FEI公司);Magna-IR560ESP型傅里葉紅外光譜儀(FTIR,美國Nicolet公司);6520型液相色譜質譜(LC-MS)分析儀(美國AB公司);GW-RO型超純水器(北京普析通用儀器有限責任公司)。
1.2 藥品與試劑
樺木酸原料藥(陜西慧科生物科技有限公司,批號:111802-201001,純度:98%);無水乙醇(天津市天力化學試劑有限公司,批號:20130408,分析純);甲醇(江蘇漢邦科技有限公司,批號:131583,色譜純);溴化鉀(常州市海拓實驗儀器有限公司,批號:20160421,光譜純);水為去離子水。
2 方法與結果
2.1 反溶劑重結晶法制備樺木酸納米粒
利用一些有機溶劑對樺木酸進行溶解度測試后,選擇乙醇作為溶劑,將一定質量的樺木酸原料藥與一定體積的乙醇溶液放入燒杯中,置于恒溫水浴鍋中并在恒溫磁力攪拌器下控制其攪拌的速度,使原料藥充分溶解,得到樺木酸乙醇溶液。待溫度穩定后,將一定體積的反溶劑(去離子水)緩慢逐滴加入到樺木酸乙醇溶液中,得到樺木酸納米混懸液。收集樺木酸納米混懸液,8 000 r/min離心(離心半徑:10 cm)10 min,棄上清,取一部分沉淀用等體積的去離子水復溶,通過Zeta電位及粒度分析儀測得粒徑大小;另一部分沉淀于-40℃中預凍12 h,再冷凍48 h,即得樺木酸納米粒。
2.2 單因素試驗篩選處方工藝
本研究在預試驗基礎上篩選了5個對樺木酸納米粒的形態和粒徑影響較大的因素,分別為樺木酸乙醇溶液質量濃度、反溶劑-溶劑體積比、攪拌速度、反溶劑滴加速度和反應溫度。每個因素設4個水平:樺木酸乙醇溶液質量濃度為0.5、1、2、3、4、5 mg/mL;反溶劑-溶劑體積比為0.5∶1、1∶1、2∶1、3∶1、4∶1、5∶1;攪拌速度為600、700、800、900、1 000、1 100 r/min;反溶劑滴加速度為2、4、6、8、10、12 mL/min;反應溫度為10、15、20、25、30、35℃。分別考察上述因素對樺木酸納米粒的影響,以粒徑最小原則進行篩選,每組試驗重復3次。
2.1.1 樺木酸乙醇溶液質量濃度在磁力攪拌速度為900 r/min、反應溫度為25℃的條件下,按反溶劑-溶劑體積比4∶1將去離子水以8 mL/min的滴加速度分別加入到質量濃度為1、2、3、4 mg/mL的樺木酸乙醇溶液中,制成樺木酸納米粒,測定其粒徑。結果顯示,當質量濃度為3 mg/mL時所制樺木酸納米粒的粒徑最小,故確定樺木酸乙醇溶液質量濃度為3 mg/mL,詳見圖1。
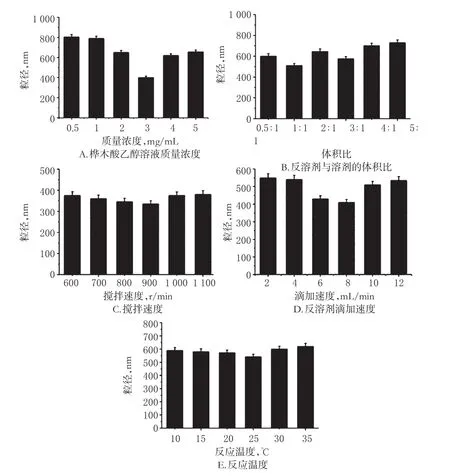
圖1 單因素試驗結果Fig1 Results of single factor test
2.1.2 反溶劑-溶劑體積比在磁力攪拌速度為900 r/min、反應溫度為25℃的條件下,按反溶劑-溶劑體積比分別為1∶1、2∶1、3∶1、4∶1將去離子水以8 mL/min的滴加速度分別加入到質量濃度為3 mg/mL的樺木酸乙醇溶液中,制成樺木酸納米粒,測定其粒徑。結果顯示,當反溶劑-溶劑體積比為1∶1時所制樺木酸納米粒的粒徑最小,故確定反溶劑-溶劑體積比為1∶1,詳見圖1。
2.1.3 攪拌速度在磁力攪拌速度分別為700、800、900、1 000 r/min,反應溫度為25℃的條件下,按反溶劑-溶劑體積比為1∶1將去離子水以8 mL/min的滴加速度加入到質量濃度為3 mg/mL的樺木酸乙醇溶液中,制成樺木酸納米粒,測定其粒徑。結果顯示,攪拌速度為900 r/min時所制樺木酸納米粒的粒徑最小,故確定攪拌速度為900 r/min,詳見圖1。
2.1.4 反溶劑滴加速度在磁力攪拌速度為900 r/min、反應溫度為25℃的條件下,按反溶劑-溶劑體積比為1∶1將去離子水以4、6、8、10 mL/min的滴加速度分別加入到質量濃度為3 mg/mL的樺木酸乙醇溶液中,制成樺木酸納米粒,測定其粒徑。結果顯示,反溶劑滴加速度為8 mL/min時所制樺木酸納米粒的粒徑最小,故確定反溶劑滴加速度為8 mL/min,詳見圖1。
2.1.5 反應溫度在磁力攪拌速度為900 r/min,反應溫度分別為15、20、25、30℃的條件下,按反溶劑-溶劑體積比為1∶1將去離子水以8 mL/min的滴加速度加入到質量濃度為3 mg/mL的樺木酸乙醇溶液中,制成樺木酸納米粒,測定其粒徑。結果顯示,反應溫度為25℃時所制樺木酸納米粒的粒徑最小,故確定反應溫度為25℃,詳見圖1。
綜上單因素試驗結果,樺木酸納米粒的處方工藝條件為:樺木酸乙醇溶液質量濃度為3 mg/mL,反溶劑-溶劑體積比為1∶1,攪拌速度為900 r/min,反溶劑滴加速度為8 mL/min,反應溫度為25℃。
2.3 正交試驗優化處方工藝
2.3.1 正交試驗在單因素試驗結果的基礎上,確定以樺木酸乙醇溶液質量濃度(A,mg/mL)、反溶劑-溶劑體積比(B)、反溶劑滴加速度(C,mL/min)和反應溫度(D,℃)為因素,按L16(45)表設計正交試驗,每個因素設4個水平:A為1、2、3、4 mg/mL,B為1∶1、2∶1、3∶1、4∶1,C為4、6、8、10 mL/min,D為15、20、25、30℃。以納米混懸液粒徑為指標,以粒徑最小原則進行篩選,每組試驗重復試驗3次,采用正交試驗優化樺木酸納米粒處方工藝。因素與水平見表1,正交試驗設計與結果見表2,方差分析結果見表3。
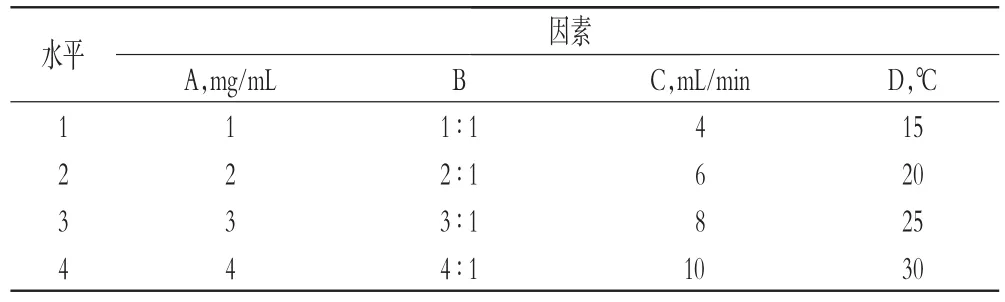
表1 因素與水平Tab1 Factors and levels
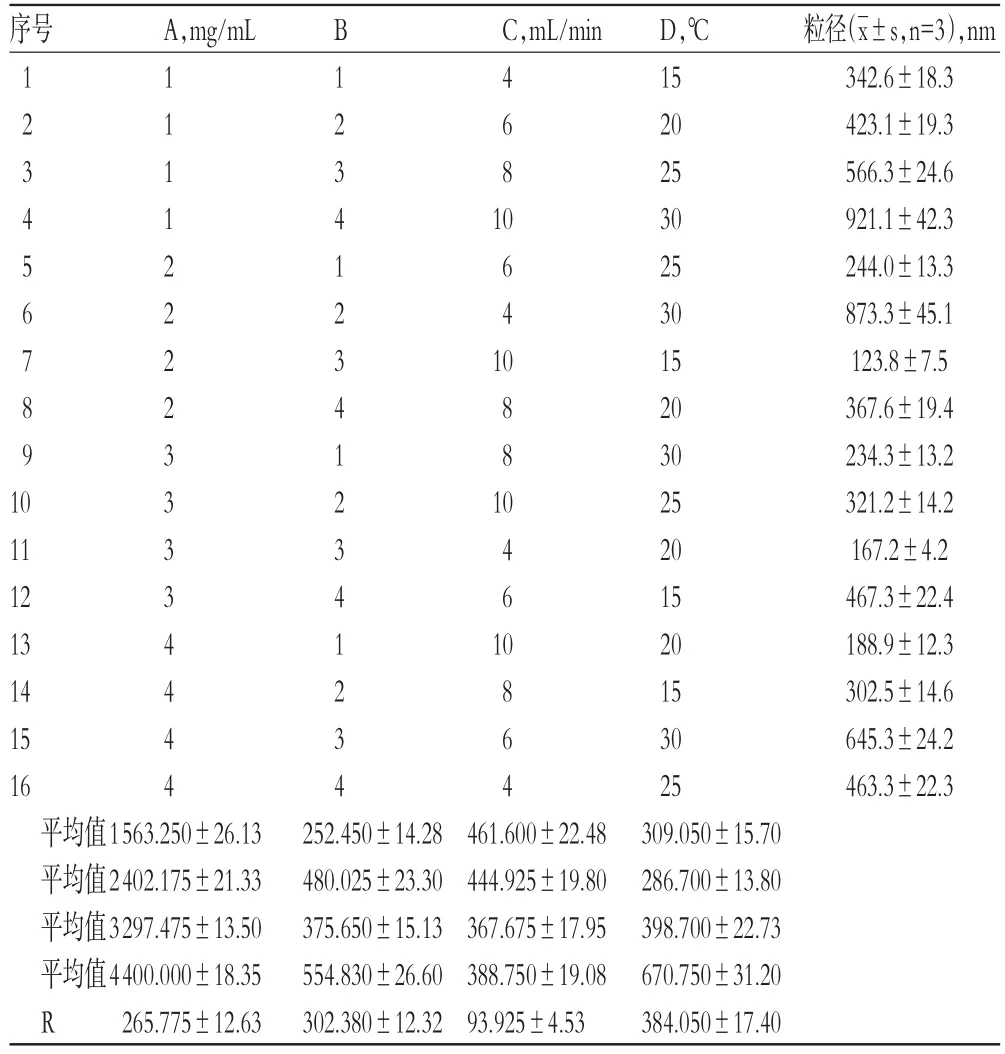
表2 正交試驗設計與結果Tab2 Orthogonal test design and results
由表2和表3結果可知,各因素對樺木酸納米粒粒徑大小的影響程度依次為反應溫度>反溶劑-溶劑體積比>樺木酸乙醇溶液質量濃度>反溶劑滴加速度,其中反溶劑-溶劑體積比、反應溫度、樺木酸乙醇溶液質量濃度對粒徑有顯著影響(P<0.05或P<0.01)。綜合考慮,確定木酸納米粒的最優處方工藝條件為A3B1C3D2,即樺木酸乙醇溶液質量濃度為3 mg/mL,反溶劑-溶劑體積比為1∶1,反溶劑滴加速度為8 mL/min,反應溫度為20℃。

表3 方差分析結果Tab3 Results of variance analysis
2.3.2 驗證試驗按最優處方工藝條件,首先稱取30 mg的樺木酸原料藥溶于10 mL的乙醇中,充分溶解得到質量濃度為3 mg/mL的樺木酸乙醇溶液;在20℃溫度下,向樺木酸乙醇溶液中以8 mL/min的滴加速度加入反溶劑(去離子水)10 mL,即制備出樺木酸納米混懸液;混懸液離心后凍干制成樺木酸納米粒。試驗重復3次并測定所制樺木酸納米粒的粒徑。結果顯示,樺木酸納米混懸液平均粒徑為(156.0±8.6)nm(n=3),凍干后粒徑為(235.0±12.2)nm(n=3)。
2.4 樺木酸納米粒表征
2.4.1 SEM分析用SEM分析最優處方工藝所制樺木酸納米粒和樺木酸原料藥的形貌和大小。操作步驟:樣品臺上黏好雙面銅導電膠,在此導電膠上黏附少量樣品,再對樣品噴金處理(電流為3 mA、時間為6 min或電流為50 mA、時間為30 s),將噴金處理后的樣品置于樣品室中觀察。結果顯示,樺木酸原料藥呈針狀的晶體結構,平均粒徑約在3 000~20 000 nm之間,粒度分布不均勻;樺木酸納米粒呈近球形,大小比較均勻,形態較規整,平均粒徑約為300 nm。樺木酸原料藥和樺木酸納米粒的SEM圖見圖2。

圖2 樺木酸原料藥和樺木酸納米粒的SEM圖Fig2 SEM pictures of betulinic acid raw material and nanoparticles
2.4.2 FTIR分析用FTIR分析最優處方工藝所制樺木酸納米粒和樺木酸原料藥的化學結構。操作步驟:將樣品與溴化鉀粉末按1∶100混合,研成細粉末、壓片,進行紅外光譜分析。結果顯示,樺木酸納米粒和樺木酸原料藥的紅外吸收峰位置基本相同,說明樺木酸納米粒與樺木酸原料藥對比,其化學結構沒有發生改變。樺木酸的紅外特征峰為3 522 cm-1為羧基的O—H伸縮振動吸收峰,1 760 cm-1為羧基的C=O伸縮振動吸收峰,1 082、1 035 cm-1為C—O伸縮振動吸收峰。樺木酸原料藥和樺木酸納米粒的紅外光譜圖見圖3。
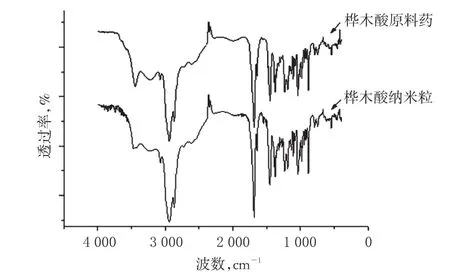
圖3 樺木酸原料藥和樺木酸納米粒的紅外光譜圖Fig3 IR pictures of betulinic acid raw material and nanoparticles
2.4.3 LC-MS分析采用LC-MS分析儀對最優處方工藝所制樺木酸納米粒和樺木酸原料藥的相對分子量進行分析。操作步驟:將樣品5 mg溶于10 mL甲醇溶液中,以10 000 r/min離心(離心半徑:10 cm)10 min,取上清進行檢測。結果顯示,樺木酸納米粒的相對分子量與樺木酸原料藥比較未發生明顯變化,樺木酸納米粒分子質子化之后的質荷比為456.3,這一結果與樺木酸分子式計算出來的結果(分子量為456.7)吻合。兩種不同晶型的樺木酸具有相同的分子量,這一結果與FTIR結果吻合,由此判斷出通過反溶劑重結晶法制得的樺木酸納米粒的化學結構沒有發生變化。樺木酸原料藥和樺木酸納米粒的LC-MS圖見圖4。
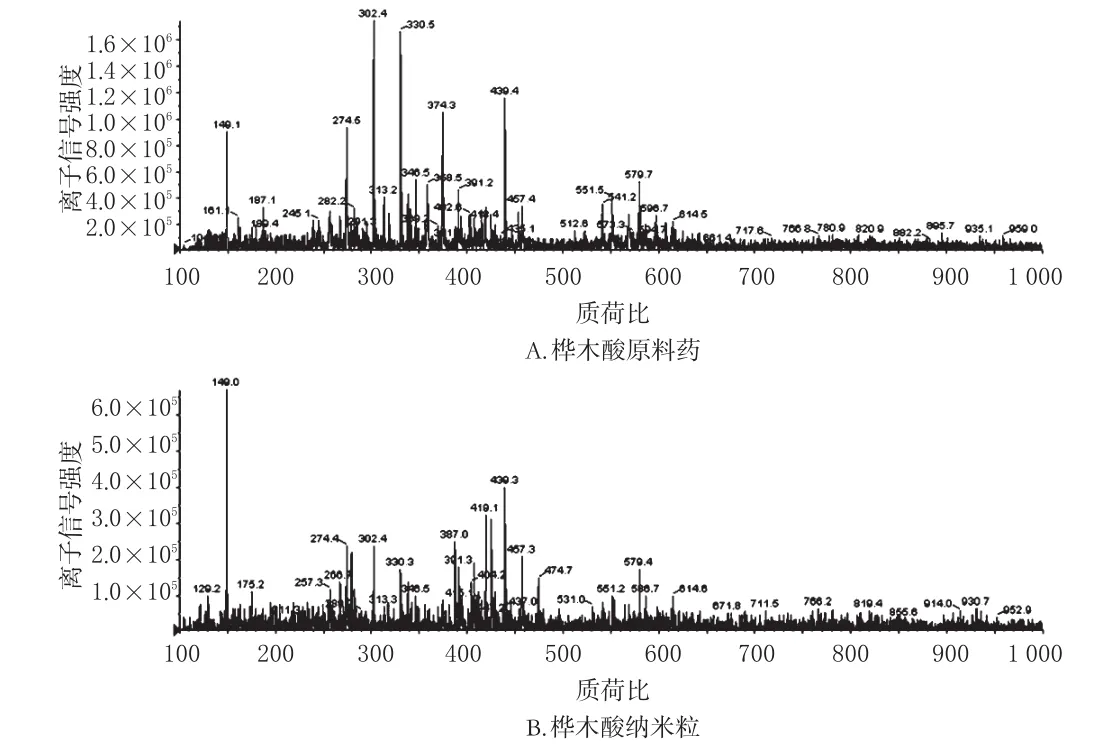
圖4 樺木酸原料藥和樺木酸納米粒的LC-MS圖Fig4 LC-MS pictures of betulinic acid raw material and nanoparticles
3 討論
本研究通過單因素試驗和正交試驗對樺木酸納米粒的處方工藝進行了優化。結果顯示,最優處方工藝條件為:樺木酸乙醇溶液質量濃度3 mg/mL、反溶劑-溶劑體積比1∶1、反溶劑滴加速度8 mL/min、反應溫度20℃、攪拌速度900 r/min。所制得的樺木酸納米混懸液粒徑為(156.0±8.6)nm(n=3),凍干后粒徑為(235.0±12.2)nm(n=3)。
本試驗以難溶于水的藥物樺木酸為研究對象,考察了反溶劑重結晶法制備樺木酸納米粒的可行性,并對所制納米粒進行了表征,為進一步開發樺木酸新劑型的研究奠定了良好的基礎。
[1] 胡祥正,馮亞亞.樺木酸及其衍生物的制備與抗腫瘤作用的研究進展[J].化學通報,2016,79(7):589-596.
[2] 易金娥,鄔靜,文利新,等.樺木酸的藥理作用研究進展[J].中草藥,2014,45(14):2118-2124.
[3] 徐先祥,秦思,李瓊.樺木酸抗炎作用與機制[J].中國現代應用藥學,2014,31(10):1284-1287.
[4] 朱利娟,向思亭,王喜紅,等.樺木酸抗炎機制研究進展[J].中獸醫醫藥雜志,2016,35(3):18-22.
[5] 鄭萍.極禿普梭木中某些化合物的抗瘧和抑制膽堿酯酶的作用[J].現代藥物與臨床,2009,24(2):113-114.
[6] Liu Y,Gao D,Zhang X,et al.Antitumor drug effect of betulinic acid mediated by polyethylene glycol modified liposomes[J].Mater Sci Eng C Mater Biol Appl,2016,doi:10.1016/j.msec.2016.03.080.
[7] 秦軍,顧曉娟.藥物超細微粒制備技術的概述[J].海峽藥學,2013,25(1):8-10.
[8] 張維,張志云,張志麗.超臨界流體技術制備固體分散體的研究進展[J].安徽醫藥,2013,17(6):903-905.
[9] de Paiva Lacerda S,Espitalier F,Hoffart V,et al.Liquid anti-solvent recrystallization to enhance dissolution of CRS 74,a new antiretroviral drug[J].Drug Dev Ind Ph-arm,2015,41(11):1910-1920.
[10] 姜茹,祖元剛,趙修華,等.超臨界反溶劑法制備葉酸介導吡柔比星葡聚糖納米粒的工藝研究[J].中國藥房,2011,22(21):1961-1964.
Preparation and Characterization of Betulinic Acid Nanoparticles
WU Mingfang,XU Wenjia,ZU Yuangang,ZHAO Xiuhua(Key Laboratory of Forest Plant Ecology,Ministry of Education,Northeast Forestry University,Harbin 150040,China)
OBJECTIVE:To prepare the betulinic acid nanoparticles,and characterize them.METHODS:Using ethanol as solvent and water as anti-solvent,anti-solvent recrystallization method was used to prepare betulinic acid nanoparticles.Using particle size as indicator,single factor test and orthogonal test were adopted to optimize the mass concentration of betulinic acid solution,anti-solventsolvent volume ratio,anti-solvent drip rate,reaction temperature and stirring speed in formulation technology of betulinic acid nanoparticles,and verification test was conducted.The betulinic acid nanoparticles were characterized by scanning electron microscopy,laser particle size analyzer,Fourier infrared spectrometer and mass spectrum analyzer.RESULTS:The optimal technology was as follow as betulinic acid solution mass concentration of 3 mg/mL,anti-solvent-solvent volume ratio of 1∶1,anti-solvent drip rate of 8 mL/min,reaction temperature of 20℃and stirring speed of 900 r/min.The average size of prepared betulinic acid nanosuspension was(156.0±8.6)nm(n=3)and the particle size was(235.0±12.2)nm(n=3)after freeze-drying,with nearly spherical appearance,uniform size and regular form.Compared with raw material of betulinic acid,the chemical structure of prepared betulinic acid nanoparticles did not change,and there were no significant changes in molecular weight and mass ratio.CONCLUSIONS:Betulinic acid nanoparticles are successfully prepared.
Betulinic acid;Nanoparticles;Anti-solvent recrystallization method;Single factor test;Orthogonal test;Characterization
R943
A
1001-0408(2017)31-4445-04
DOI 10.6039/j.issn.1001-0408.2017.31.32
*博士研究生。研究方向:植物化學與植物藥。E-mail:604533864@qq.com
#通信作者:教授,博士。研究方向:藥物新劑型。E-mail:xiuhuazhao@nefu.edu.cn
2017-03-25
2017-08-17)
(編輯:鄒麗娟)
