愈創木酚甘油醚和右美沙芬復方制劑中雜質同時測定的方法研究
2017-09-08 09:57:31柴洪帆
中國藥業 2017年16期
關鍵詞:檢測
柴洪帆,鄭 揚,曹 霞
(海南澳美華制藥有限公司,海南 海口 570311)
·檢驗檢測·
愈創木酚甘油醚和右美沙芬復方制劑中雜質同時測定的方法研究
柴洪帆,鄭 揚,曹 霞
(海南澳美華制藥有限公司,海南 海口 570311)
目的 建立可同時檢測愈創木酚甘油醚和右美沙芬復方制劑中有關物質的方法。方法 色譜柱為Waters XBridge C18柱(250 mm× 4.6 mm,3.5 m),流動相A為pH=3.0的磷酸二氫鉀溶液-乙腈(90∶10,V/V),流動相B為pH=2.8的磷酸二氫鉀溶液-乙腈-甲醇(10∶10∶80,V/V/V),梯度洗脫,檢測波長為280 nm。結果 強制降解條件(氧化、酸、堿、高溫、光照)下,主成分與雜質均得到了有效檢測。愈創木酚甘油醚、右美沙芬和其他8種特定雜質質量濃度在線性范圍內與峰面積線性關系良好。結論 所建立的方法準確度、檢測限、定量限、精密度、耐用性等均符合人用藥品注冊技術要求國際協調會議(ICH)指導原則的要求。
反相高效液相色譜法;愈創木酚甘油醚;右美沙芬;復方制劑;雜質
右美沙芬(dextromethorphan,DM)為 3-甲氧基-17-甲基-(9α,13α,14α)-嗎啡喃氫溴酸一水合物,為白色或類白色結晶性粉末,無臭[1]。愈創木酚甘油醚(guaifenesin,GG)為 3-(2-甲氧基苯氧基)丙烷-1,2-二醇,口服后刺激胃黏膜,反射性引起支氣管分泌增加,使痰液稀釋[2]。二者的復方制劑臨床主要用于治療急、慢性支氣管炎和流行性感冒引起的咳嗽、痰多、咽痛等[3],也常用于兒童咳嗽的治療[4]。有關物質的檢測是復方制劑質量控制中最具挑戰性的任務。愈創木酚甘油醚和氫溴酸右美沙芬收載于美國藥典(USP35)和歐洲藥典(EP8.0),復方制劑未見收載。目前,單一組分的檢測方法得到了廣泛研究[5-8],但關于右美沙芬和愈創木酚甘油醚復方制劑的質量研究較少[9-11]。本研究的主要目的是建立愈創木酚甘油醚和右美沙芬復方制劑的有關物質測定方法,用于監控產品的化學雜質,保證產品的安全性和有效性。現報道如下。
1 儀器與試藥
1.1 儀器
高效液相色譜儀,包括Waters Alliance E2695分離系統、Waters 2998光電二極管陣列檢測器、柱溫箱及 Empower 3色譜工作站;Waters XBridge C18柱(250 mm×4.6 mm,3.5 μm);Binder 720P型藥物穩定性試驗箱;XS 105DU型十萬分之一電子天平,XP26型百萬分之一電子天平(MettlerToledo公司)。
1.2 試藥
甲醇(色譜純,FisherScientific);氫溴酸右美沙芬對照品(DM,批號為100201-201204),愈創木酚甘油醚對照品(GG,批號為100528-201303),均購自中國食品藥品檢定研究院;氫溴酸右美沙芬雜質A對照品(DM-A,批號為Y0000261,EP);氫溴酸右美沙芬雜質B對照品(DM-B,批號為492,LGC);氫溴酸右美沙芬雜質C對照品(DM-C,批號為35952,LGC);氫溴酸右美沙芬雜質氮氧化物(DM-N-Oxide,批號為 32441,LGC);愈創木酚甘油醚雜質A對照品(GG-A,批號為C4X-1294-1510,CATO);愈創木酚甘油醚雜質B對照品(GG-B,批號為21182,LGC);愈創木酚甘油醚雜質C對照品(GG-C,批號為52038,LGC);愈創木酚甘油醚雜質 D對照品(GG-D,批號為 CX4-1294-1601,CATO);復方氨酚美沙芬口服液(批號為16071401,16072201,16072202,自制樣品);其他試劑均為分析純。各雜質化學名及簡寫見表1。

表1 愈創木酚甘油醚和右美沙芬雜質的化學名及簡寫
2 方法與結果
2.1 溶液制備
系統適用性溶液:分別精密稱取愈創木酚甘油醚雜質對照品(GG-A,GG-B,GG-C,GG-D),氫溴酸右美沙芬雜質對照品(DM-A,DM-B,DM-C,DM-NOxide),氫溴酸右美沙芬對照品和愈創木酚甘油醚對照品,分別配制成約含愈創木酚甘油醚雜質對照品0.003 g/L,氫溴酸右美沙芬雜質對照品0.02 g/L,GG 0.6 g/L及DM 4 g/L的溶液,作為系統適用性溶液。
供試品溶液:精密吸取復方氨酚美沙芬口服液2.4 g,置10 mL容量瓶中,加入甲醇-水(50∶50)溶液適量,超聲30 min,定容至刻度,濾過,取續濾液,作為供試品溶液。
2.2 色譜條件優選
2.2.1 檢測波長
采用全波長檢測,分別對主成分DM和GG及其他8種已知雜質進行測定,結果全部組分在215~230 nm及273~304 nm波長范圍有最大吸收,由于215~230 nm波長接近末端吸收,最終選擇280 nm作為檢測波長。試驗結果顯示,在此檢測波長下,主成分及其他雜質均有良好吸收,同時基線較為平穩。特定雜質紫外吸收光譜圖見圖1。
2.2.2 流動相梯度洗脫條件
起始梯度洗脫條件的流動相:流動相A為緩沖鹽-乙腈(90∶10,V∶V),流動相B為緩沖鹽-乙腈-甲醇(10∶10∶80,V∶V∶V);緩沖鹽為0.01 mol/L的磷酸二氫鈉和0.004 6 mol/L的正辛烷磺酸鈉水溶液,稀磷酸調節pH為3.0,梯度洗脫條件見表2。分別考察不同的色譜條件下,系統適用性試驗中各雜質峰和主峰,以及雜質峰之間的分離情況,結果顯示,由于DM-A,DM,DM-N-Oxide極性極為相似,色譜圖中雜質峰與主峰重疊而無法分離(見圖2)。通過后續的梯度洗脫條件、緩沖鹽溶液濃度和種類、柱溫等優化篩選得到了最終梯度洗脫條件(見表2),系統適用性溶液中主峰和8個雜質峰可達到分離要求。

圖1 特定雜質的紫外吸收光譜圖

表2 流動相優選過程中代表性梯度洗脫條件
梯度洗脫起始條件(階段1)篩選:通過篩選起始條件,調整主成分在適當的時間出峰,更有利于主成分與其他8個特定雜質的分離。通過階段1起始流動相比例調整和梯度斜率的變化,最終選擇起始流動相A與B比例由85∶15變化為92∶8,使主成分GG的保留時間由初始的約10 min調整至約20 min,相應DM的保留時間由21 min調整至35~38 min。結果最終選定流動相A與B的比例為92∶8作為啟動梯度條件。后續進一步考察DM及與其極性相似的組分(DM-A,DM-NOxide)的分離情況。

圖2 優化前色譜條件下系統適用性試驗色譜圖
梯度變化階段條件(階段2和階段3)篩選:在設定階段2條件流動相比例為50∶50時分離效果不理想的情況下,保持梯度比例斜率不變,進一步考察流動相A與B的比例由50∶50變化為40∶60(即梯度5和梯度17)的分離效果,初步確定采用40∶60更有利于DMA,DM,DM-N-Oxide的分離。進一步篩選階段3梯度條件的變化(梯度11至梯度17),結果顯示,流動相A比例升高有助于各成分分離,階段3等度洗脫時效果更佳,進一步增加流動相A比例后更有利于DM-A,DM,DM-N-Oxide,DM-A,GG-C的分離。篩選過程中具有代表性的梯度條件參見圖3。

圖3 梯度洗脫條件篩選
梯度條件優化:等度洗脫無法達到完全分離,因此變更為梯度洗脫,變化流動相A的比例,同時為達到全部雜質能盡快出峰而得到檢測,提高階段4的有機相比例,從而得到優化的最終梯度洗脫條件(見表2)。采用優化的梯度條件進行檢測,全部組分達到了分離度要求(圖4)。梯度條件優化過程中具有代表性的梯度條件見圖5。

圖4 最終選定色譜條件下系統適用性試驗色譜圖

圖5 梯度洗脫條件優化
2.2.3 流動相緩沖鹽溶液
緩沖鹽溶液組成:分別采用不同的磷酸鹽(磷酸二氫鈉、磷酸二氫鉀)配制緩沖鹽溶液并配制流動相,依法進行檢測。結果顯示,不同種類的鹽對吸收強度有影響,配制相同濃度的緩沖鹽,采用磷酸二氫鉀作為流動相時雜質的響應值更高,更有利于方法的靈敏度。最終選擇磷酸二氫鉀配制緩沖鹽溶液。
緩沖鹽溶液濃度:分別采用磷酸二氫鉀配制不同濃度的緩沖鹽溶液并配制流動相,依法進行檢測。結果顯示,緩沖鹽濃度對峰形有影響。最終選擇磷酸二氫鉀濃度為0.01 mol/L和正辛烷磺酸鈉濃度為0.004 6 mol/L。
緩沖鹽溶液pH:分別調節緩沖鹽溶液的不同pH,進一步配制成流動相,依法進行檢測。結果顯示,不同pH的流動相對基線均有一定的影響,pH=2.8時基線最平穩,更有利于方法的靈敏度。最終選擇緩沖鹽溶液的pH=2.8。
2.2.4 樣品稀釋劑
分別采用不同的稀釋劑制備供試品加樣溶液(加入限度水平的雜質),依法檢測。結果顯示,由于不同成分(主成分或雜質)的化學穩定性不同,稀釋劑體系會影響供試品溶液的穩定性,采用甲醇-水溶液(50∶50)為稀釋劑時穩定性最佳,能保證供試品加樣溶液在24 h內穩定。
2.2.5 色譜柱
所建立的檢測方法需同時測定10種成分,其中3種成分極性與DM極為相似,為保證方法的適用性,進行了色譜柱的篩選。分別對品牌、型號、常規參數等不同的色譜柱行比較試驗,最終選擇Waters XBridge C18柱(250 mm×4.6 mm,3.5 μm)。
2.2.6 柱溫
在45~55℃范圍內對柱溫進行考察,結果隨著溫度的升高,主峰與雜質峰的分離度增大,柱溫為50℃時分離度良好。
2.3 初步擬訂的檢測色譜條件
色譜柱:Waters XBridge C18柱(250 mm×4.6 mm,3.5μm);流動相:流動相A為緩沖鹽溶液-乙腈(90∶10),流動相B為緩沖鹽溶液-乙腈-甲醇(10∶10∶80),緩沖鹽溶液為0.01 mol/L的磷酸二氫鈉和0.004 6 mol/L的正辛烷磺酸鈉水溶液,稀磷酸調節pH為2.8,梯度洗脫,洗脫程序見表2的最終條件;流速:0.8 mL/min;柱溫:50℃;進樣量:20 μL;檢測波長:280 nm。
2.4 方法學考察
2.4.1 專屬性試驗
將樣品經高溫、光照、酸、堿和氧化破壞,依法測定。結果顯示,樣品在高溫及光照環境中較穩定,基本無降解;在堿性環境極不穩定,降解產物有待進一步明確。同時,在各降解條件下,DM和GG與輔料峰、各雜質峰及降解產物峰均能達到良好分離,方法專屬性良好。色譜圖見圖6。

圖6 強制降解試驗的典型圖譜
2.4.2 檢測限測定
取各雜質對照品適量,配制成母液,將母液逐步稀釋,依法進樣測定。將信噪比(S/N)為3∶1時的進樣量作為檢測限,結果見表3。
2.4.3 線性關系考察
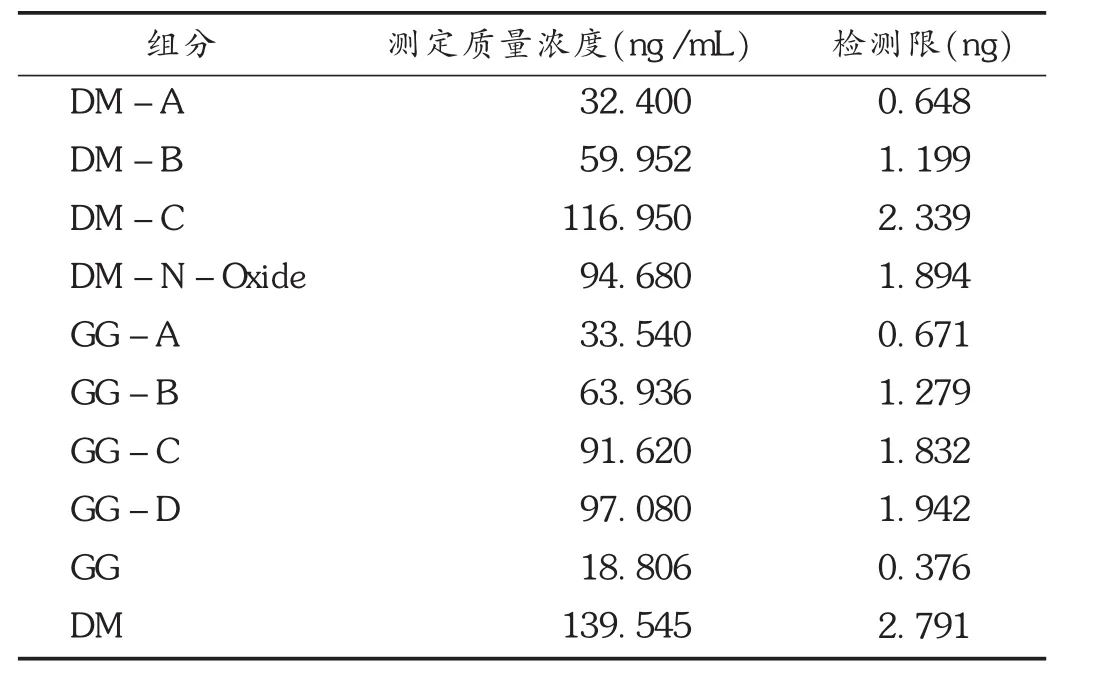
表3 檢測限測定結果
取各雜質對照品適量,配制系列對照品溶液,注入高效液相色譜儀測定,記錄色譜圖。以質量濃度(X,μg/mL)和峰面積(Y)進行線性回歸,計算得回歸方程。結果表明,主成分和各雜質對照品質量濃度在線性范圍內與峰面積線性關系良好。根據測定的標準曲線,采用標準曲線測定法進一步計算校正因子,用于雜質含量的計算。結果見表4。

表4 線性關系考察結果

表5 樣品有關物質測定結果(%)
2.4.4 精密度試驗
平行配制6份供試品溶液,依法進樣測定。結果峰面積的 RSD≤1.5%(n=6),表明儀器精密度良好。
2.4.5 穩定性試驗
取加樣供試品溶液,分別于配制0,2,4,6,8,10,12,18,20,24 h時各進樣測定1次。結果主峰峰面積的RSD為 1.6%(n=10),各限度水平的雜質峰面積的RSD≤5.0%(n=10),表明供試品溶液在室溫放置24 h內穩定性良好。
2.4.6 耐用性試驗
分別考察了不同批號色譜柱、流動相配比、流速和柱溫的微小波動對系統適用性溶液中各峰分離度的影響。結果上述條件微小變化及色譜柱批號變化后,各物質峰之間仍能達到基線分離,雜質峰之間分離度均不小于1.2,表明方法的耐用性良好。
2.5 樣品檢測
取3批樣品,依法進樣測定,以加校正因子主成分自身對照法計算有關物質的含量。結果見表5。
3 討論
經查詢文獻,右美沙芬復方制劑的法定標準或藥典標準皆未收載其有關物質檢查方法,因此,建立其有關物質檢測與控制方法對于產品的質量控制具有重要意義。右美沙芬復方制劑中,氫溴酸右美沙芬和愈創木酚甘油醚的穩定性具有差異,同時規格相差較大(氫溴酸右美沙芬為3 g/mL,愈創木酚甘油醚為20 g/L),為保證質量,需要明確各雜質的歸屬并進行控制,且需要檢驗方法具有較高的專屬性和靈敏度。
復方制劑中主成分氫溴酸右美沙芬和愈創木酚甘油醚極性相差較大,DM-C,DM,DM-N-Oxide,DM-A和GG-C極性相似,為有關物質方法的研究帶來了挑戰。采用高效液相色譜法分析,能夠實現較好的分離效果。因此,本研究中采用反相高效液相色譜梯度洗脫條件進行設計,分別對梯度洗脫條件、緩沖鹽的種類和溶液濃度、柱溫、色譜柱等色譜條件進行了篩選。結果表明,在擬訂色譜條件下,主成分DM和GG、防腐劑苯甲酸鈉(BN)、DM和GG的8個已知雜質,以及其他雜質均可達到良好分離,從而建立了行之有效的有關物質檢測方法,可用于產品的質量控制[12]。進一步對檢測方法進行方法學考察,結果表明,方法的準確度、檢測限、定量限、精密度、耐用性等均符合ICH指導原則的要求。
[1]Papich MG.Dextromethorphan.Saunders Handbook of Veterinary Drugs(Fourth Edition):Small and Large Animal[M].Elsevier LTD:Oxford,2016:225-226.
[2]Sherrington LA,Sherrington A.Guaifenesin[J].Analytical Profilesof Drug Substances and Excipients,1998,25:121-164.
[3]Paul IM,Yoder KE,Crowell KR,et al.Effect of dextromethorphan,diphenhydramine,and placebo on nocturnal cough and sleep quality for coughing children and their parents[J].Pediatrics,2004,114(1):85-90.
[4]Yoder KE,Shaffer ML,La Tournous SJ,et al.Child assessment of dextromethorphan,diphenhydramine,and placebo for nocturnalcough due to upper respiratory infection[J].Clin Pediatr(Phila),2006,45(7):633-640.
[5]Amaratunga P,Clothier M,Lorenz Lemberg B,et al.Determination of Dextromethorphan in Oral Fluid by LC-MS-MS[J].J Anal Toxicol,2016,40(5):360-366.
[6]Xu R,Xu T,Wang Z,et al.Simultaneous determination of dextromethorphan and dextrophan in rat plasma by LC-MS/MS and its application to a pharmacokinetic study[J].Pharmazie,2012,67(6):485-489.
[7]Reddy PS,Sudhakar Babu K,Kumar N,et al.Development and validation of stability indicating the RP-HPLC method for the estimation of related compounds of guaifenesin in pharmaceutical dosageforms[J].PharmaceuticalMethods,2011,2(4):229-234.
[8]Grosa G,Del Grosso E,Russo R,et al.Simultaneous,stability indicating,HPLC-DAD determination of guaifenesin and methyl and propyl-parabens in cough syrup[J].Journal of Pharmaceutical and Biomedical Analysis,2006,41(3):798-803.
[9]Eichhold TH,McCauley-Myers DL,Khambe DA,et al.Simultaneous determination of dextromethorphan,dextrorphan,and guaifenesin in human plasma using semi-automated liquid/liquid extraction and gradient liquid chromatography tandem mass spectrometry[J].Pharm Biomed Anal,2007,43(2):586-600.
[10]Raju R,Kumar NA,Kumar SR.Development and Validation of a Stability-Indicating RP-HPLC Method for the Simultaneous Estimation of Guaifenesin and Dextromethorphan Impurities in PharmaceuticalFormulations[J].Chromatography Research International,2013:315145.
[11]McSharry WO,Savage IV.Simultaneous high-pressure liquid chromatographic determination of acetaminophen,guaifenesin,and dextromethorphan hydrobromide in cough syrup[J].Journal of Pharmaceutical Sciences,1980,69(2):212-214.
[12]Rajagopalan R.Review of regulatory guidance on impurities[J].Separation Science and Technology,2004,5:27-37.
Simultaneous Determination of Impurities in Guaifenesin and Dextromethorphan Compound Preparation
Chai Hongfan,Zheng Yang,Cao Xia
(Hainan Bright Future Pharmaceutical Co.,Ltd.,Haikou,Hainan,China 570311)
Objective To establish a method for simultaneous determination of guaifenesin and dextromethorphan compound preparation.Methods The chromatographic column was Waters XBridge C18column(250 mm×4.6 mm,3.5μm).The mobile phase A consisted of pH=3.0 monopotassium phosphate solution-acetonitrile(90∶10,V/V),the mobile phase B consisted of pH=3.0 monopotassium phosphate solution-acetonitrile-methanol(10∶10∶80,V/V/V),gradient elution,the detection wavelength was 280 nm.Results Under the conditions of forced degradation(oxidation,acid,alkali,high temperature,light),the principal components and impurities were effectively detected.Guaifenesin,dextromethorphan and other 8 kinds of specific impurities had a good linear relationship in the concentration range.Conclusion The established method is validated according to ICH guidelines with respect to accuracy,limits of inspection,limits of quantification,precision and durability.
RP-HPLC;guaifenesin;dextromethorphan;compound preparation;impurity
R927
A
1006-4931(2017)16-0018-06
2016-12-07;
2017-04-28)
10.3969/j.issn.1006-4931.2017.16.006
柴洪帆,女,碩士研究生,中級工程師,研究方向為藥品質量控制,(電子信箱)wangy1580@163.com。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
