烯胺酮(酯)參與的串聯(lián)反應構(gòu)建吡咯衍生物的研究進展
2017-08-29 23:50:36傅鴻樑吳逸冰鐘為慧浙江工業(yè)大學藥學院長三角綠色制藥協(xié)同創(chuàng)新中心浙江杭州310014
合成化學 2017年8期
傅鴻樑, 王 輝, 吳逸冰, 凌 飛, 鐘為慧(浙江工業(yè)大學 藥學院 長三角綠色制藥協(xié)同創(chuàng)新中心,浙江 杭州 310014)
·綜合評述·
烯胺酮(酯)參與的串聯(lián)反應構(gòu)建吡咯衍生物的研究進展
傅鴻樑, 王 輝, 吳逸冰, 凌 飛, 鐘為慧*
(浙江工業(yè)大學 藥學院 長三角綠色制藥協(xié)同創(chuàng)新中心,浙江 杭州 310014)
作為多位點的有機合成子,烯胺酮(酯)參與的串聯(lián)反應廣泛應用于吡咯衍生物的合成。綜述了烯胺酮(酯)與鄰二羰基化合物,β-硝基烯烴和苯乙炔等參與的串聯(lián)反應在吡咯單環(huán)和稠環(huán)衍生物合成中的應用進展,并對其未來發(fā)展進行了展望。參考文獻49篇。
烯胺酮(酯); 串聯(lián)反應; 吡咯衍生物; 研究進展; 綜述
串聯(lián)反應是將多步反應或多個反應組分置于同一個反應容器中進行,第一步生成的中間體無需分離提純,可直接進行下一步反應得到最終產(chǎn)物。串聯(lián)反應具有原子經(jīng)濟性、環(huán)境友好和易于操作等特點。隨著社會經(jīng)濟快速發(fā)展,環(huán)境保護問題日益嚴峻,人們對“綠色化學”與“原子經(jīng)濟性”日益重視。串聯(lián)反應作為多樣性導向合成的有效方法,成功應用于不對稱反應和雜環(huán)分子合成,在構(gòu)建復雜分子[1-2],特別是構(gòu)建具有光學活性的天然產(chǎn)物和稠雜環(huán)骨架[3]中有廣泛應用。
烯胺酮(酯)是經(jīng)典的有機化合物之一,最早由Greenhill[4]提出。該類化合物結(jié)構(gòu)中含有N—C=C—Z(Z=COR, CO2R)共軛結(jié)構(gòu),也稱為β-氨基-α,β-不飽和酮(酯)。從結(jié)構(gòu)上看,烯胺酮(酯)兼具烯胺的親核性和烯酮的親電性,能發(fā)生烯醇-酮式互變和烯胺-亞胺互變,可以作為1,3-偶極子參與1,3-偶極環(huán)加成反應[5],也可與α,β-不飽和化合物進行Michael加成、環(huán)合等串聯(lián)反應[6]。由于烯胺酮(酯)具備作為多位點合成子的功能,合成工作者深入研究了其理化性質(zhì)[7]和在合成雜環(huán)化合物中的應用[8-9]。
2006年,李明等[10]詳細分析了烯胺酮的4種可能構(gòu)象,并綜述了烯胺酮經(jīng)典合成方法及其在單雜環(huán)化合物合成中的應用。2014年,萬結(jié)平等[11]綜述了基于N,N-二甲基烯胺酮與不同胺基親核試劑發(fā)生交叉胺化過程的多組分串聯(lián)反應,重點介紹了其在1,4-二氫吡啶、吡啶等雜環(huán)合成中的應用進展。2016年,Wan等[12]綜述了N,N-二甲基烯胺酮結(jié)構(gòu)中C—N鍵交叉胺化、C=C雙鍵打開和原位生成烯胺酮參與串聯(lián)反應的研究進展。
雖然烯胺酮(酯)在串聯(lián)反應中的研究已取得了諸多成果,但其參與的串聯(lián)反應在合成雜環(huán)母體結(jié)構(gòu)多樣性方面的應用仍然有限,現(xiàn)有綜述鮮有其用于吡咯骨架的報道。已有串聯(lián)反應構(gòu)建吡咯衍生物的綜述,主要以烯胺酮(酯)作為關鍵中間體形式出現(xiàn),如1,3-二羰基類、丙炔酸酯類化合物與苯胺等直接形成烯胺酮(酯),繼而與相應原料進行環(huán)合得到吡咯骨架。在產(chǎn)物結(jié)構(gòu)上,以吡咯單環(huán)為主。近年來,雖然逐漸出現(xiàn)了烯胺酮(酯)烯丙位C(sp3)—H鍵活化、分子內(nèi)串聯(lián)環(huán)化、與炔烴串聯(lián)氧化環(huán)化等反應的報道,但尚沒有相關文獻總結(jié)烯胺酮(酯)直接參與的該類反應在稠環(huán)吡咯衍生物構(gòu)建中的應用。因此,本文從反應底物的類型出發(fā),綜述了烯胺酮(酯)與鄰二羰基化合物,β-硝基烯烴和苯乙炔等參與的串聯(lián)反應在吡咯單環(huán)和稠環(huán)衍生物合成中的應用進展,并對其未來發(fā)展進行了展望。
1 與鄰二羰基化合物的串聯(lián)反應
1.1 烯胺酮(酯)與苯甲酰甲醛的串聯(lián)反應
烯胺酮(酯)作為重要的有機合成砌塊,常應用于雜環(huán)(如吡啶[13]、嘧啶[14]、呋喃[15]、吡唑[16]等)的合成。吡咯環(huán)作為最常見的五元雜環(huán)之一,廣泛存在于Atorvastain, Corrole, Zomepirac和Sunitinib等天然產(chǎn)物和藥物分子結(jié)構(gòu)中(Chart 1)。吡咯骨架構(gòu)建一直是合成化學的研究熱點,經(jīng)典的Paal-Knorr和Hantzsch吡咯合成方法持續(xù)受到化學工作者的關注和改進。烯胺酮(酯)通過與各種鄰二羰基化合物,如苯甲酰甲醛、靛紅、茚三酮等串聯(lián)反應合成吡咯衍生物。這些鄰二羰基化合物具有獨特的鄰位羰基結(jié)構(gòu),可與各種親核試劑反應構(gòu)建不同的雜環(huán)骨架。
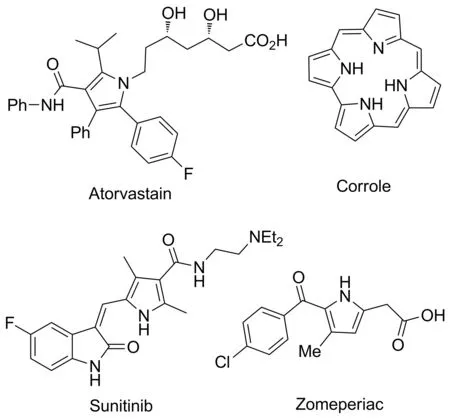
Chart 1
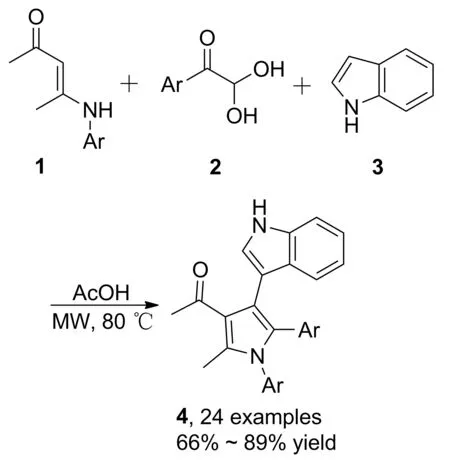
烯胺酮(酯)參與串聯(lián)反應用于吡咯單環(huán)合成的報道相對較少。2013年,Tu等[17]報道了一種在微波輔助和醋酸催化下,烯胺酮、苯甲酰甲醛、吲哚參與的串聯(lián)反應,合成了多取代吡咯衍生物(4, Scheme 1),收率66%~89%。
Scheme 1
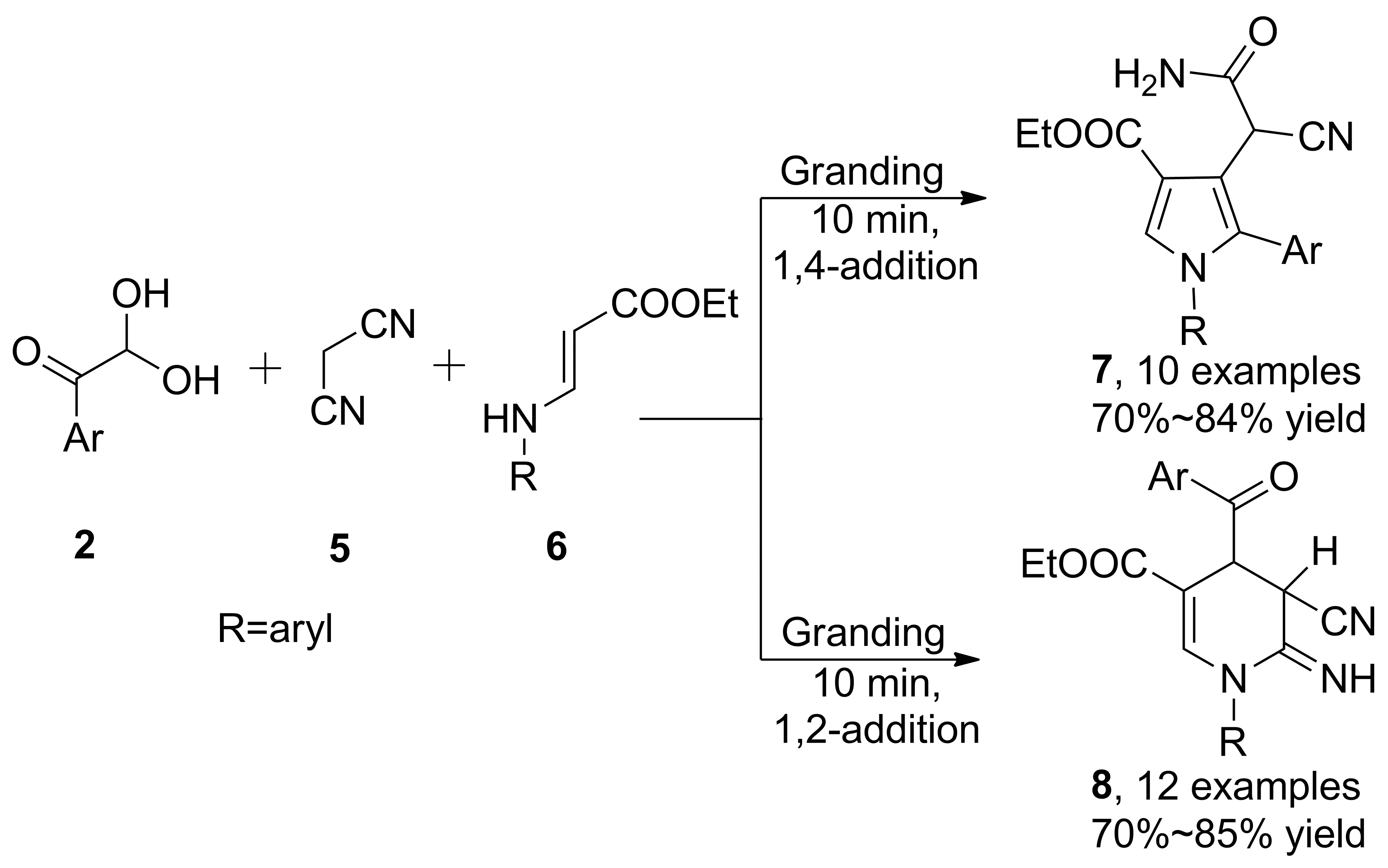
2016年,Padmini等[18]報道了苯甲酰甲醛、丙二腈、烯胺酯的三組分串聯(lián)反應(Scheme 2),研磨條件下不加入溶劑和催化劑,依次經(jīng)Knoevenagel反應、Michael加成和分子內(nèi)環(huán)化,10 min內(nèi)完成反應得到吡咯衍生物(7),收率70%~84%。值得注意的是,烯胺酯取代基R為對位取代的芳基時,傾向于得到四氫吡啶衍生物(8),收率70%~85%,沒有檢測到吡咯環(huán)產(chǎn)物。
Scheme 2
Scheme 3
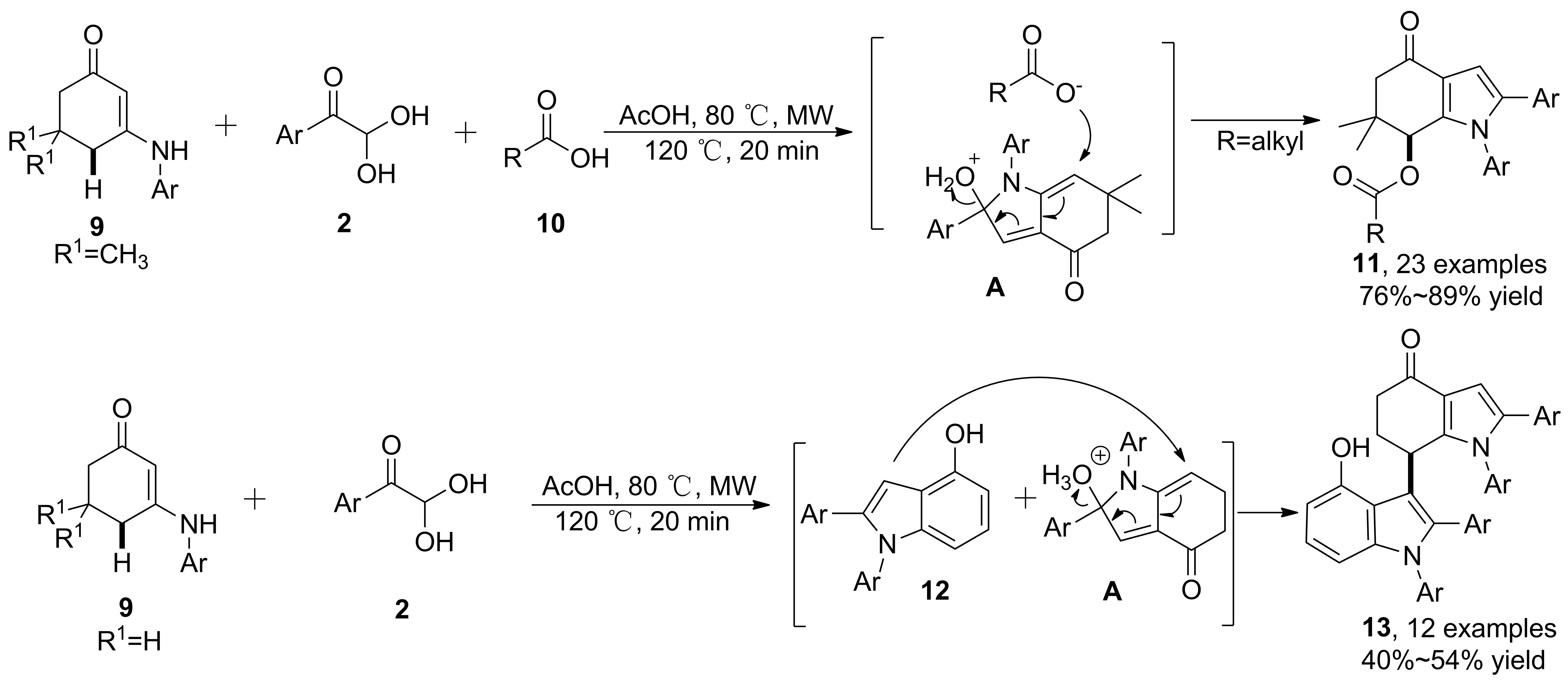
報道較多的是吡咯稠環(huán)的合成。2012年,Tu等[19]報道了環(huán)烯胺酮、苯甲酰甲醛和醋酸的三組分反應合成吡咯稠環(huán)衍生物(11)的方法。在微波輔助下,20~30 min完成反應,實現(xiàn)烯丙位C(sp3)—H鍵活化。醋酸既作為溶劑和質(zhì)子源,又作為親核試劑參與反應。根據(jù)烯胺酮底物R1結(jié)構(gòu)的不同,分別得到吡咯稠環(huán)和雙吲哚環(huán)的產(chǎn)物。可能的反應機理為:首先,烯胺酮的碳作為第一親核位點,進攻酸催化活化的苯甲酰甲醛;繼而發(fā)生脫水縮合、亞胺異構(gòu)化以及分子內(nèi)環(huán)化得到中間體(A); A與乙酸反應得到吡咯稠環(huán)(11),收率76%~89%。 R1為氫時,A可以脫水芳構(gòu)化得到吲哚中間體(12); 12與A反應得到雙吲哚骨架(13, Scheme 3),收率40%~54%。
由于醋酸的親核性相對較弱,在上述反應基礎上通過加入不同的親核試劑,化學工作者開發(fā)了一系列親核試劑修飾的吡咯稠環(huán)衍生物。2012年,Tu等[20]報道了硫酚、烯胺酮、苯甲酰甲醛的三組分反應。硫酚作為親核試劑,通過控制加料順序,選擇性地合成了不同位置硫酚取代吡咯稠環(huán)。同年,Tu等[21]用苯胺替代醋酸,通過控制環(huán)烯胺N上取代基的大小,選擇性地得到吲哚環(huán)3-位或7-位取代產(chǎn)物,在拓展底物時意外實現(xiàn)了烯胺酮N-芳基化。2013年,該課題組[22]還報道了吲哚、烯胺酮、苯甲酰甲醛的三組分反應,一鍋法區(qū)域選擇性地得到C(3)-位取代的雙吲哚衍生物的同時,沒有合成吲哚C(2)-位取代產(chǎn)物。
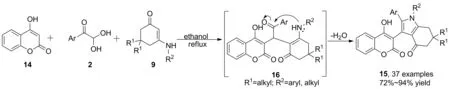
當親核試劑為活性亞甲基時,則出現(xiàn)了與上述反應機理不同的路徑。2013年,Shi等[23]報道了烯胺酮、苯甲酰甲醛、4-羥基香豆素的三組分反應。以乙醇為溶劑,在無催化劑條件下得到吡咯稠環(huán)(15),收率72%~94%。烯胺酮的烯丙位沒有出現(xiàn)C(sp3)—H鍵活化的現(xiàn)象。通過單晶衍射確證產(chǎn)物結(jié)構(gòu)后,作者提出了與Tu[19]不同的反應機理。首先,4-羥基香豆素和苯甲酰甲醛發(fā)生Knoevenagel縮合得到中間體(16); 16依次與烯胺酮發(fā)生Michael加成,亞胺烯胺互變及分子內(nèi)縮合脫水,最終得到目標產(chǎn)物(Scheme 4)。
Scheme 4
Scheme 6
1.2 與靛紅的串聯(lián)反應
靛紅早在1841年即實現(xiàn)人工合成,具有良好的生物活性,是中藥青黛的主要成分。靛紅分子內(nèi)具有鄰二羰基結(jié)構(gòu),可以提供親電位點參與串聯(lián)反應,在合成螺環(huán)化合物方面已有廣泛應用[24]。烯胺酮與靛紅參與的串聯(lián)反應用于合成吡咯衍生物報道,主要集中在吡咯并吖啶和吡咯并喹啉衍生物。2012年,Shi等[25]首次報道了L-脯氨酸催化下靛紅、烯胺酮參與的兩組分串聯(lián)反應,該反應成功構(gòu)建了吡咯[2,3,4-kI]吖啶-1-酮衍生物(Scheme 5)。在L-脯氨酸催化下活化靛紅C(3)-位羰基,經(jīng)分子內(nèi)環(huán)化、脫水后得到產(chǎn)物(18),收率78%~93%。當R為H時會發(fā)生芳構(gòu)化,得到吡咯吖啶衍生物(19),收率76%~84%。
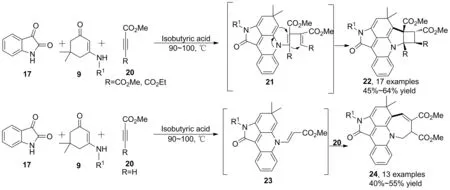
2013年,Li等[26]進一步研究了靛紅、烯胺酮以及兩分子丁炔二酸二甲酯的串聯(lián)反應。以異丁酸作催化劑,經(jīng)歷[3+2]/[4+2]/[2+2+2+1]/[2+2]環(huán)化一次性構(gòu)建4個環(huán),實現(xiàn)烯胺酮烯丙位C(sp3)—H鍵活化,得到產(chǎn)物(22),收率45%~64%。 R為H,底物為丙炔酸甲酯時,發(fā)生[5+2]環(huán)加成得到多環(huán)化合物(24, Scheme 6),收率40%~55%。
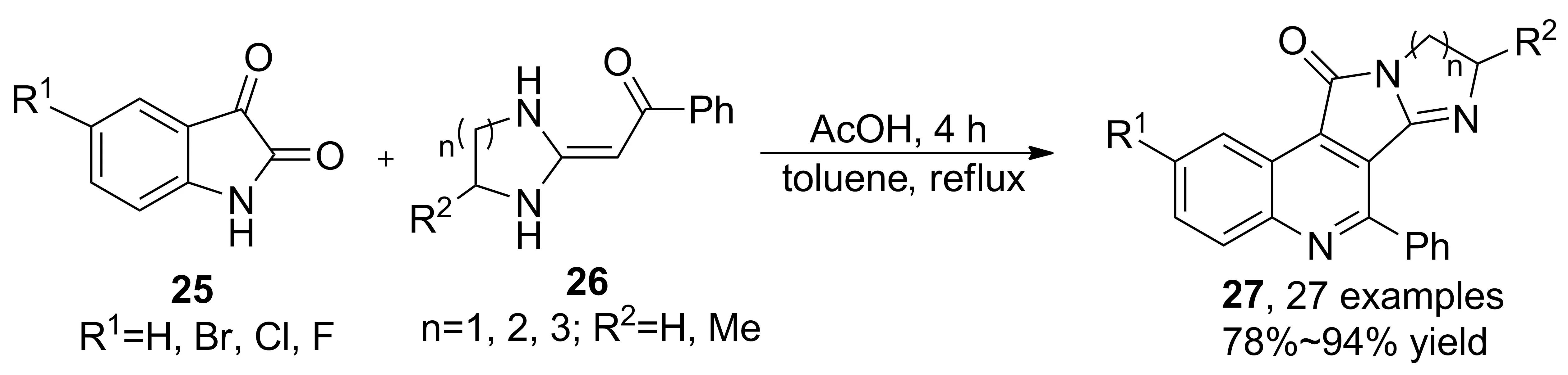
吡咯并喹啉衍生物是一類具有抗疲勞、抗炎等生物活性的分子骨架。2011年,Lin等[28]報道了烯胺酮和靛紅的串聯(lián)反應。該反應合成了咪唑并吡咯并喹啉衍生物(27, Scheme 7),首次實現(xiàn)了烯胺酮N(1), C(3)親核位點和C(1)羰基親電位點同時參與反應,收率78%~94%。烯胺酮氮雜環(huán)大小對收率影響較大,七元環(huán)收率普遍高于五元環(huán)。
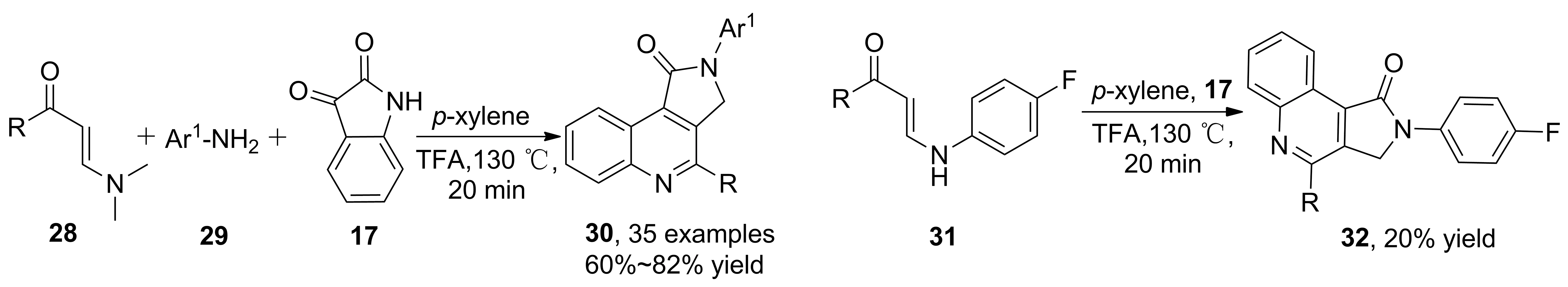
2015年,Yu等[29]報道了三氟乙酸催化下,烯胺酮、取代苯胺、靛紅三組分反應合成吡咯[3,4-c]喹啉-1-酮衍生物(30, Scheme 8)的方法,收率60%~80%。該反應的特點在于最后階段吡咯環(huán)的亞甲基碳正離子接受二甲胺給出的H-的過程。中間體(31)與靛紅反應時,排除二甲胺存在,仍能以20%收率得到產(chǎn)物。
Scheme 7
Scheme 8
Scheme 9
Scheme 10
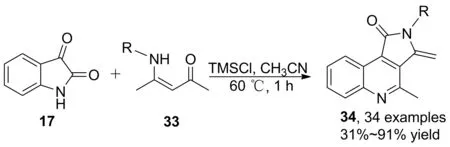
2016年,Yu等[30]報道了TMSCl催化下烯胺酮、靛紅的兩組分串聯(lián)反應,得到吡咯烷酮5-位上雙鍵修飾產(chǎn)物(34, Scheme 9)。由于雙鍵易于衍生化的特點,可以用高錳酸鉀將雙鍵氧化成羰基,進一步豐富了吡咯[3,4-c]-喹啉-1-酮衍生物的多樣性。該方法對于芳基取代的烯胺酮有較好的底物適用性,收率31%~91%,但脂肪鏈取代的烯胺酮對應的收率普遍較低。
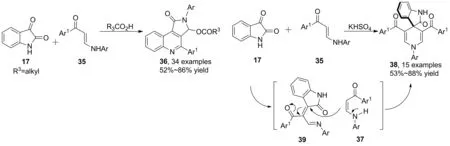
2016年,Yu等[31]報道了乙酸催化下烯胺酮、靛紅、乙酸的三組分反應,合成了吡咯[3,4-c]-喹啉-1-酮(36),收率52%~86%;用硫酸氫鉀替代乙酸催化,產(chǎn)物為螺環(huán)吲哚產(chǎn)物(38, Scheme 10),收率53%~88%。醋酸的用量可以控制兩種產(chǎn)物的比例,醋酸用量為10.0 eq時,產(chǎn)物為36。此外,其它的有機酸,如三氟乙酸、苯甲酸、對甲苯磺酸的催化效果均不如醋酸。作者認為酸的親核性和pKa值對收率有顯著影響。三氟乙酸催化時,親核性過低,導致反應產(chǎn)生的水進一步參與反應,得到吡咯酮5-位羥基取代產(chǎn)物。若溶劑為甲醇,則得到甲氧基取代產(chǎn)物。
Scheme 11
Scheme 12
Scheme 13
1.3 與茚三酮/苊醌的串聯(lián)反應
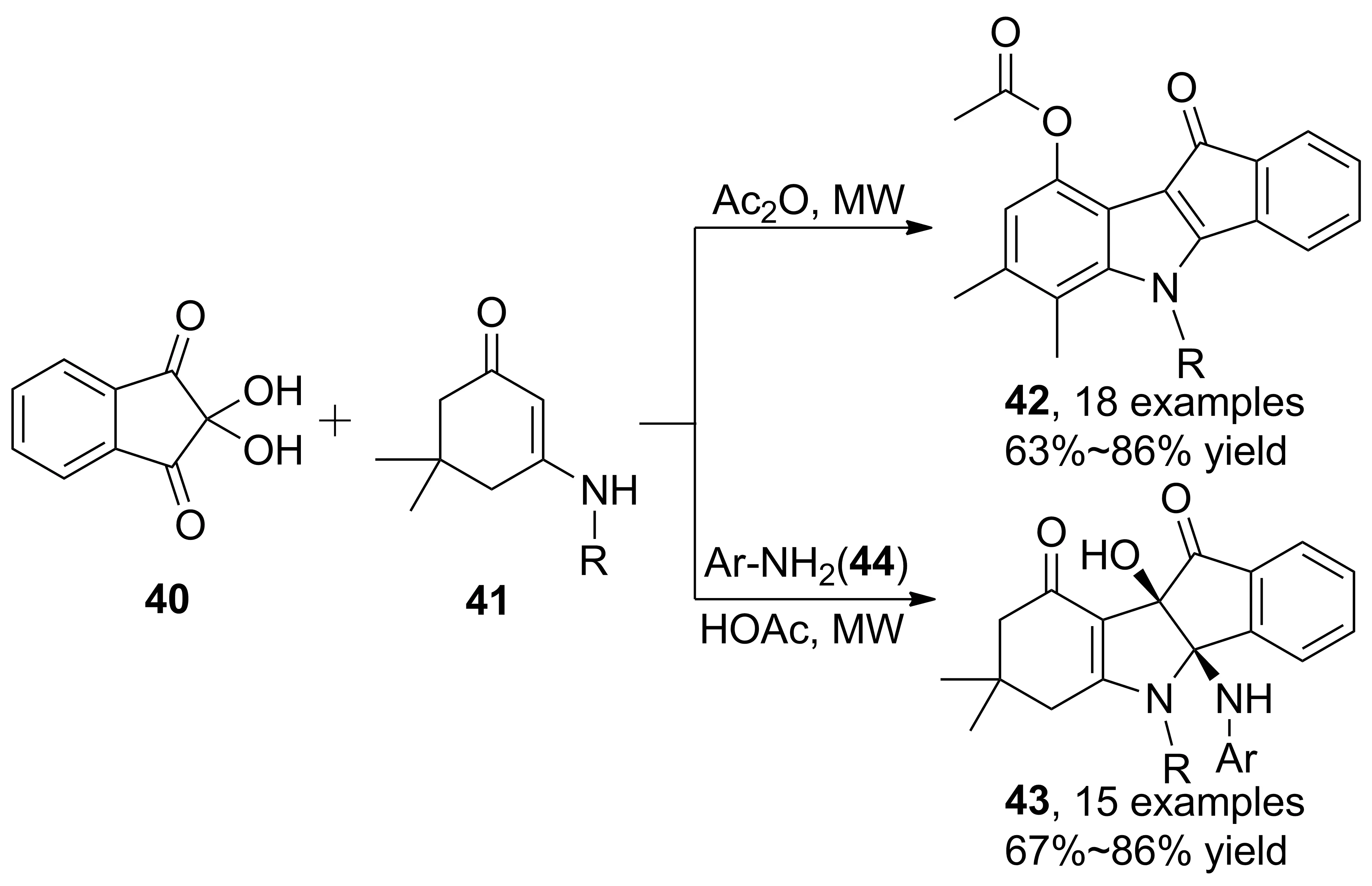
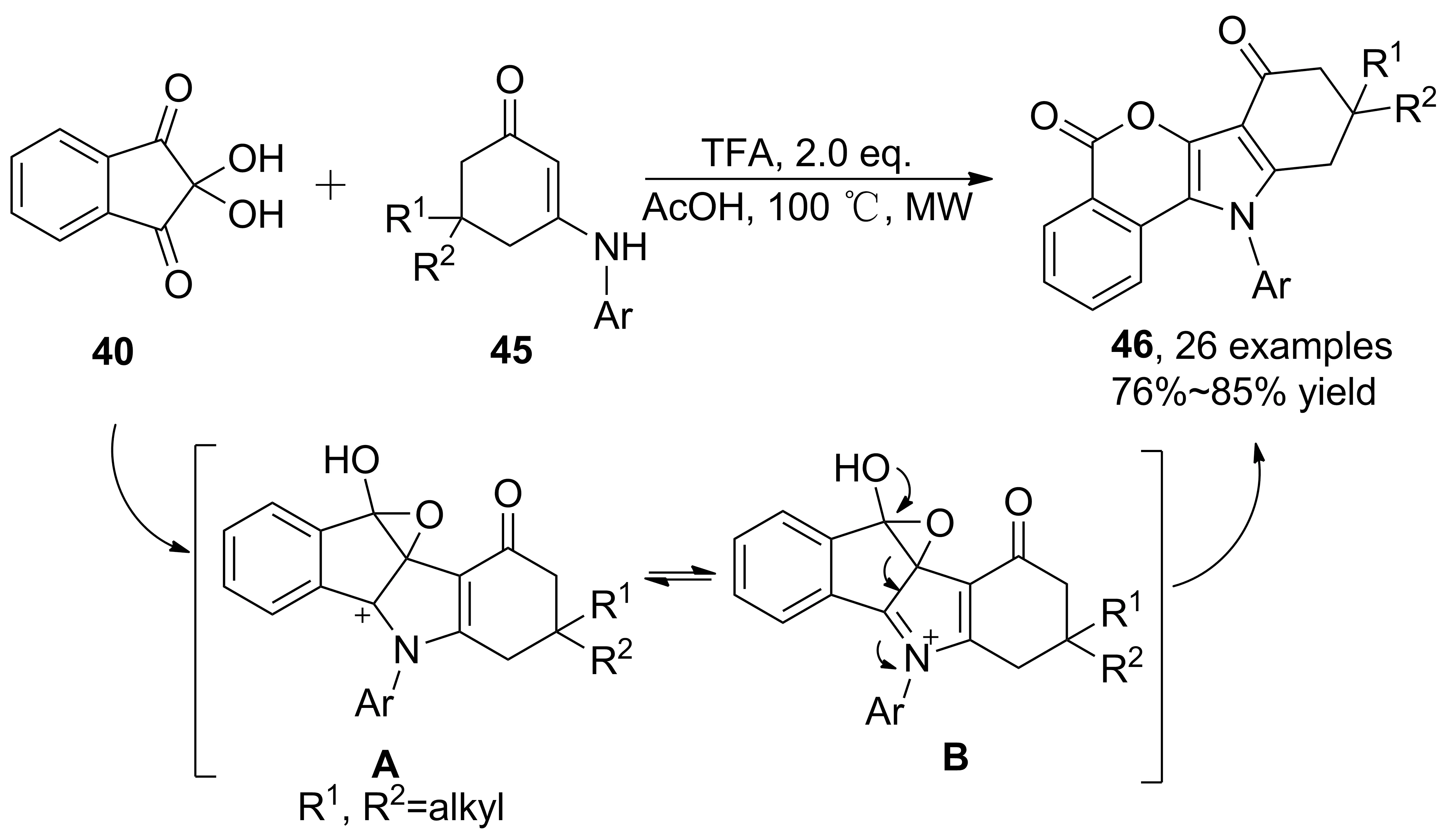
茚三酮具有與苯甲酰甲醛相似的親電中心,分子本身具有雙環(huán)結(jié)構(gòu),應用于烯胺酮的串聯(lián)反應時,可以構(gòu)建吡咯稠環(huán)衍生物。2012年,Tu等[32]報道了微波輔助下茚三酮、烯胺酮串聯(lián)反應合成茚并[1,2-b]吲哚衍生物(42, Scheme 11)。醋酸酐作為催化劑,催化活性優(yōu)于醋酸,并發(fā)生烯胺酮甲基遷移、芳構(gòu)化現(xiàn)象,沒有觀察到烯胺酮烯丙位活化后取代產(chǎn)物;在醋酸催化下芳胺作為親核試劑,則得到另一種茚并[1,2-b]吲哚衍生物43,收率67%~86%。進一步研究發(fā)現(xiàn),該串聯(lián)反應可以生成不同結(jié)構(gòu)的產(chǎn)物。 2014年,Tu等[33]采用三氟乙酸和醋酸共催化,茚三酮發(fā)生開環(huán)反應得到異色烯[4,3-b]吲哚衍生物(46, Scheme 12),收率76%~85%。
Scheme 14
Scheme 15
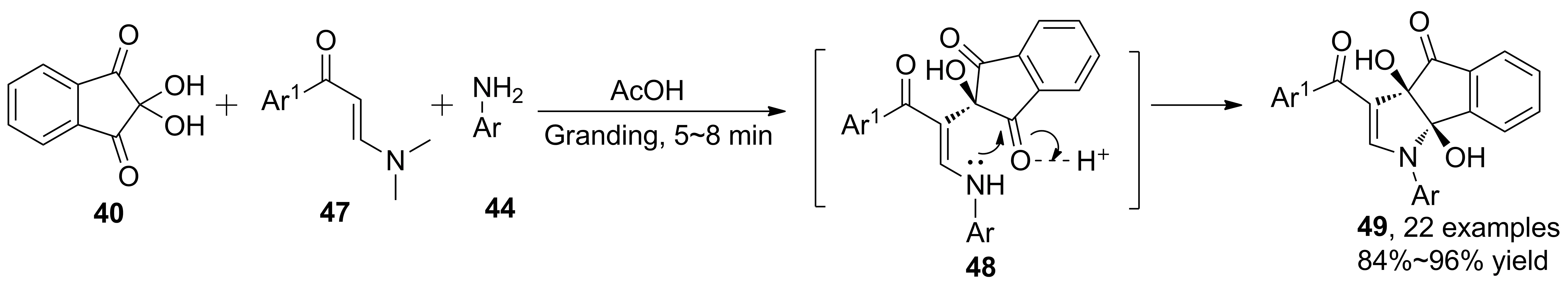
2014年,Muthusaravanan等[34]報道了研磨條件下烯胺酮參與的多組分合成茚酮并二氫吡咯衍生物的反應(49, Scheme 13)。醋酸催化下研磨5~8 min即可完成反應。結(jié)果表明,有溶劑存在時收率降低(84%~96%)。形成中間體后,受空間位阻影響,傾向于得到同側(cè)雙羥基產(chǎn)物,具有很高的區(qū)域選擇性與立體選擇性。
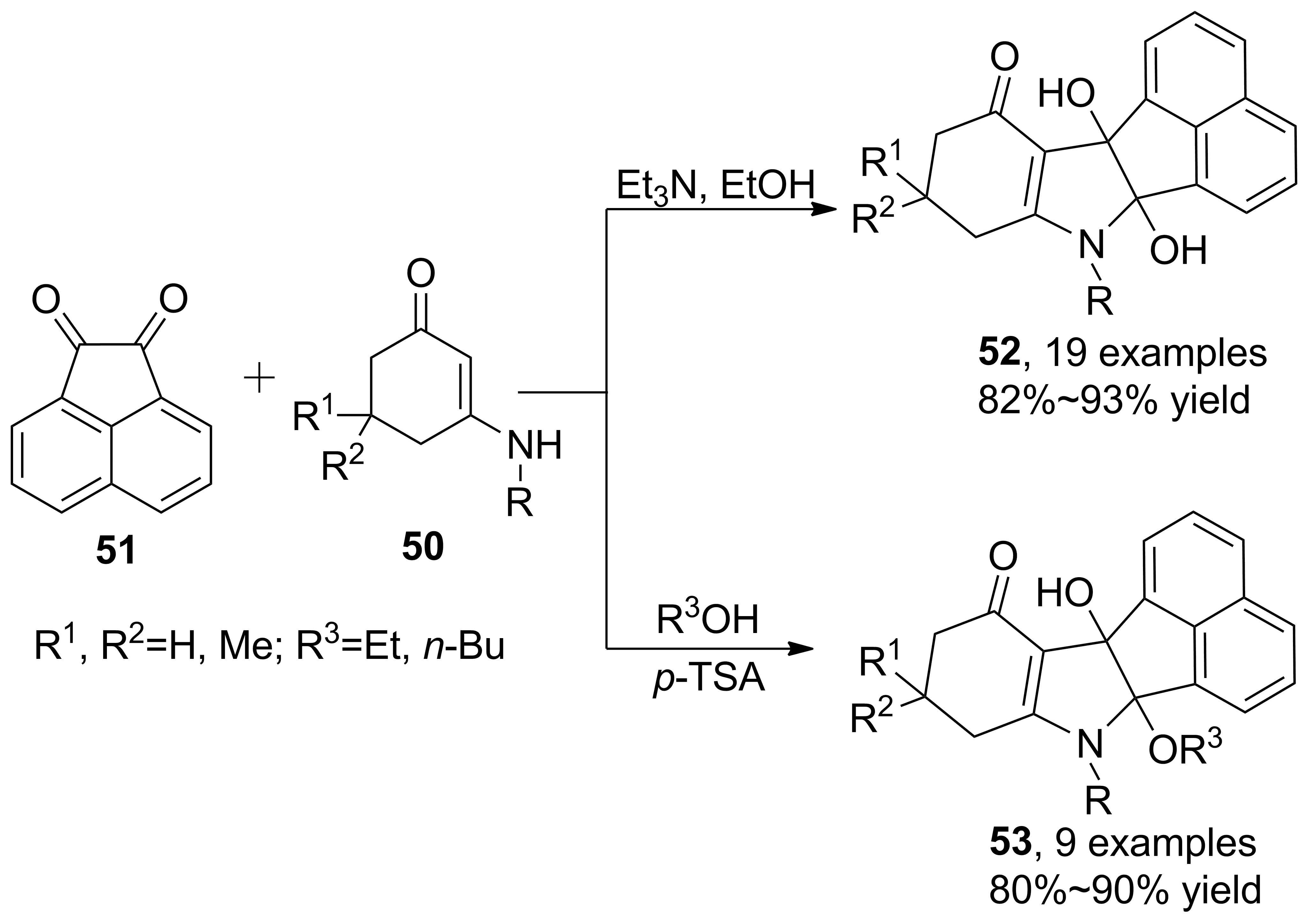
近年來,關于苊醌與烯胺酮參與的串聯(lián)反應合成吡咯衍生物也有報道。Shi等[35]開發(fā)了L-脯氨酸催化和乙醇回流條件下,烯胺酮、苊醌、巴比妥酸衍生物的三組分串聯(lián)反應,合成了四氫苊并[1,2-b]吲哚酮衍生物。反應結(jié)束后,只需經(jīng)過簡單的乙醇淋洗即可得到高純度產(chǎn)物。2015年,Lin等[36]報道了Et3N催化和乙醇回流條件下苊醌、烯胺酮的兩組分串聯(lián)反應,得到苊并[1,2-b]吲哚衍生物(52, Scheme 14),收率82%~93%。酸催化下可以繼續(xù)發(fā)生羥基質(zhì)子化脫水,醇作為親核試劑發(fā)生SN1反應,得到苊并[1,2-b]吲哚衍生物(53),收率80%~90%。
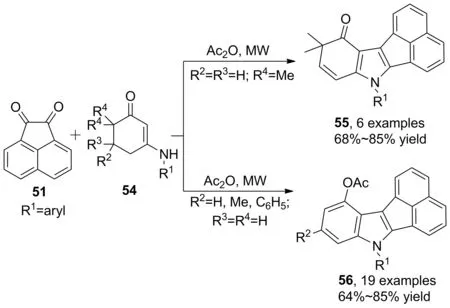
2016年,Li等[37]報道了類似的串聯(lián)反應。微波輔助,醋酸酐催化下,通過控制烯胺酮C(5)-和C(6)-位碳原子上的取代基,選擇性合成了苊并[1,2-b]吲哚衍生物(55, Scheme 15)。當R3=R4=H時,發(fā)生環(huán)烯胺酮芳構(gòu)化,得到產(chǎn)物(56),收率64%~85%。
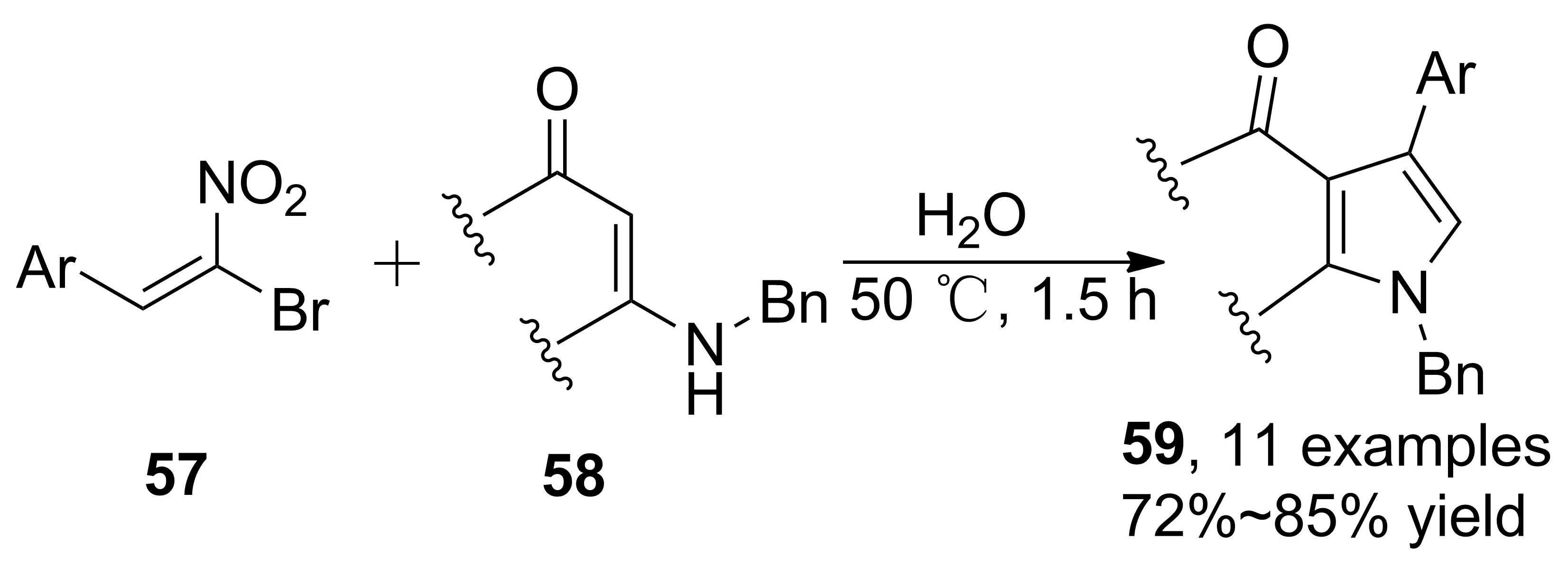
β-硝基烯烴是一類重要的合成子,硝基有很強的拉電子效應,是很好的Michael受體,并且由于硝基的易轉(zhuǎn)化性,β-硝基烯烴已經(jīng)被廣泛應用于含氮類化合物的合成。Rueping等[38]首次報道了烯胺酮、β-溴代硝基苯乙烯的串聯(lián)反應。以H2O為溶劑,無催化劑參與合成吡咯衍生物(59, Scheme 16),收率72%~85%,符合綠色化學要求。鑒于F在藥物理化性質(zhì)、藥代動力學等方面的重要作用,作者重點研究了β-溴代硝基苯乙烯芳環(huán)C(4)-位F取代底物的反應活性,結(jié)果顯示對于直鏈和環(huán)烯胺酮(酯)均有良好的適用性。
Scheme 16
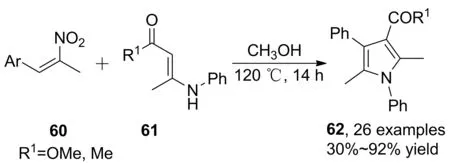
β-溴代硝基苯乙烯中由于鹵素的存在,一定程度上存在污染較大的問題。Guan等[39]報道了1-苯基-2-硝基丙烯與烯胺酯的串聯(lián)反應。在甲醇中于120 ℃反應14 h合成了多取代吡咯環(huán)化合物(62, Scheme 17),收率30%~92%。反應對烯胺酯具有良好的適用性,對烯胺酮適用范圍有限,僅有一種烯胺酮(R1=CH3)參與反應的報道,收率58%。 2013年,Guan等[40]將Blaise中間體引入1-苯基-2-硝基丙烯與烯胺酮的串聯(lián)反應,在FeCl3催化下反應20 h,成功實現(xiàn)NH吡咯環(huán)的構(gòu)建,對烯胺酯β-位上芳環(huán)取代、烷基鏈取代均有較好的適用性。
Scheme 17
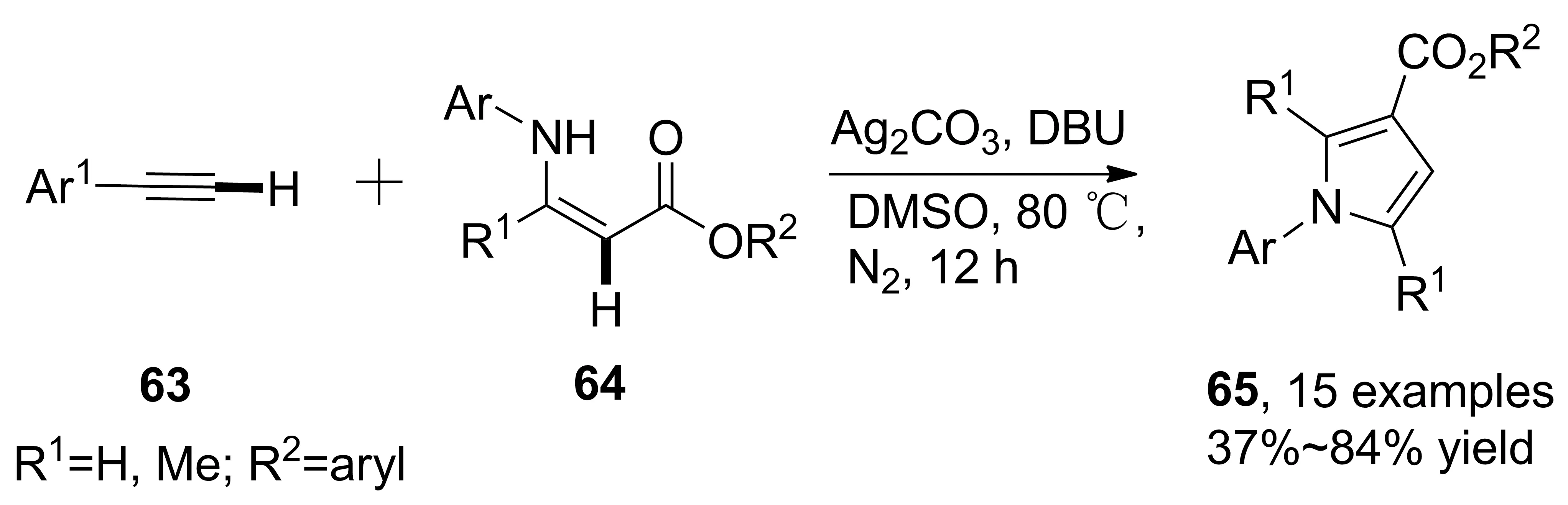
2013年,Lei等[41]以Ag2CO3和DBU共催化,烯胺酯、苯乙炔參與的串聯(lián)反應,經(jīng)歷端炔C—H鍵氧化、交叉偶聯(lián)、亞胺烯胺互變、分子內(nèi)環(huán)化合成了多取代吡咯衍生物(65, Scheme 18),收率37%~84%。該反應具有高度專一性,Ag2CO3可防止苯乙炔發(fā)生自身偶聯(lián)。
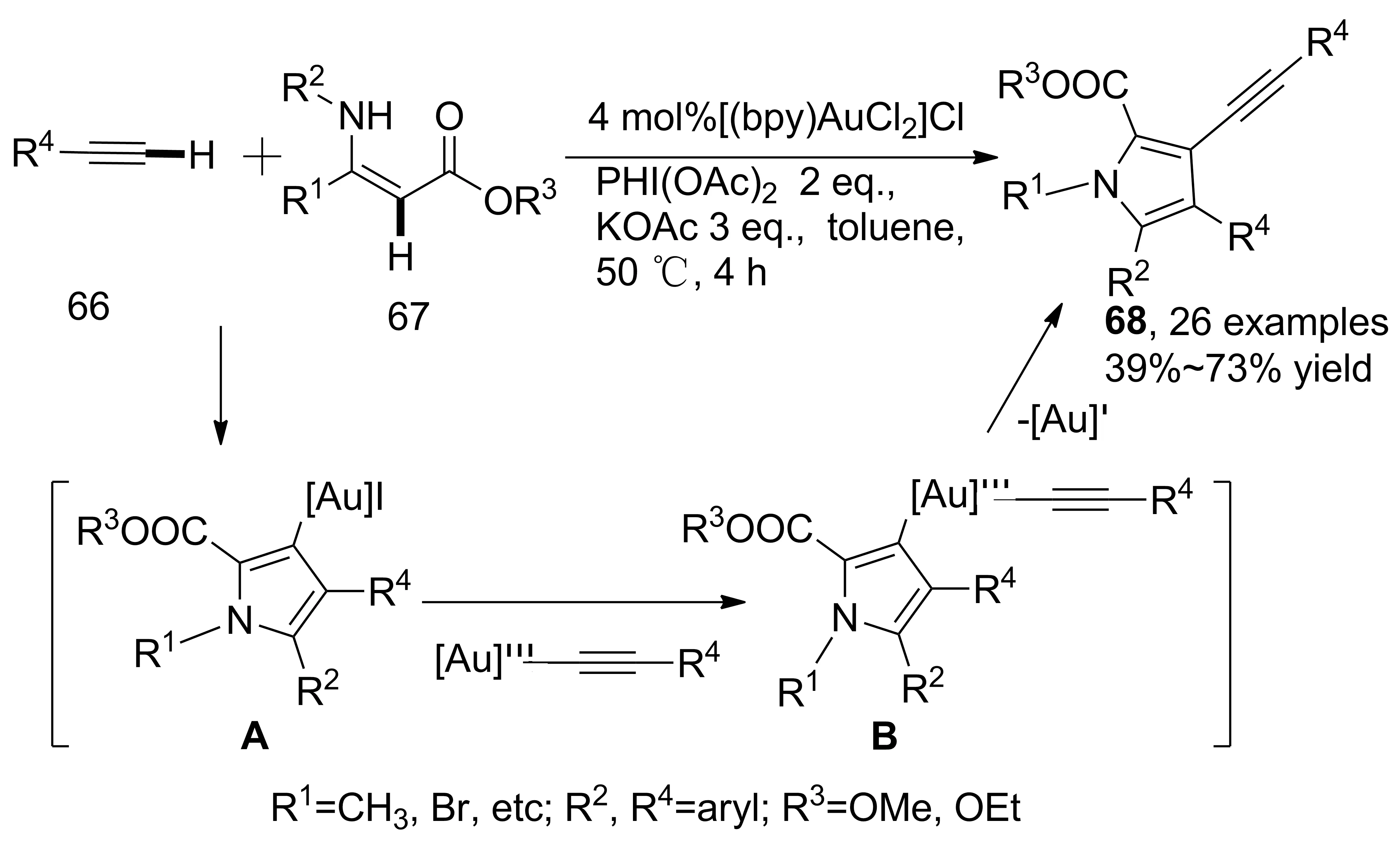
2016年,You等[42]進一步研究了金催化烯胺酯、苯乙炔參與的串聯(lián)反應(Scheme 19)。反應經(jīng)歷炔烴C—H鍵氧化、C—H鍵交叉偶聯(lián)、分子內(nèi)環(huán)化、吡咯C(3)-位炔基化等過程,在吡咯環(huán)上成功引入苯乙炔基團,收率39%~74%。機理研究表明,只有端炔才能發(fā)生反應。此外,吡咯環(huán)與苯乙炔衍生物(66)反應也沒有得到相應產(chǎn)物,表明該反應不是通過先形成吡咯環(huán),再進行吡咯C(3)-位炔烴化,而是在先生成關鍵中間體(A)后,繼續(xù)與苯乙炔偶聯(lián)形成中間體(B),再脫去一價金離子得到產(chǎn)物。
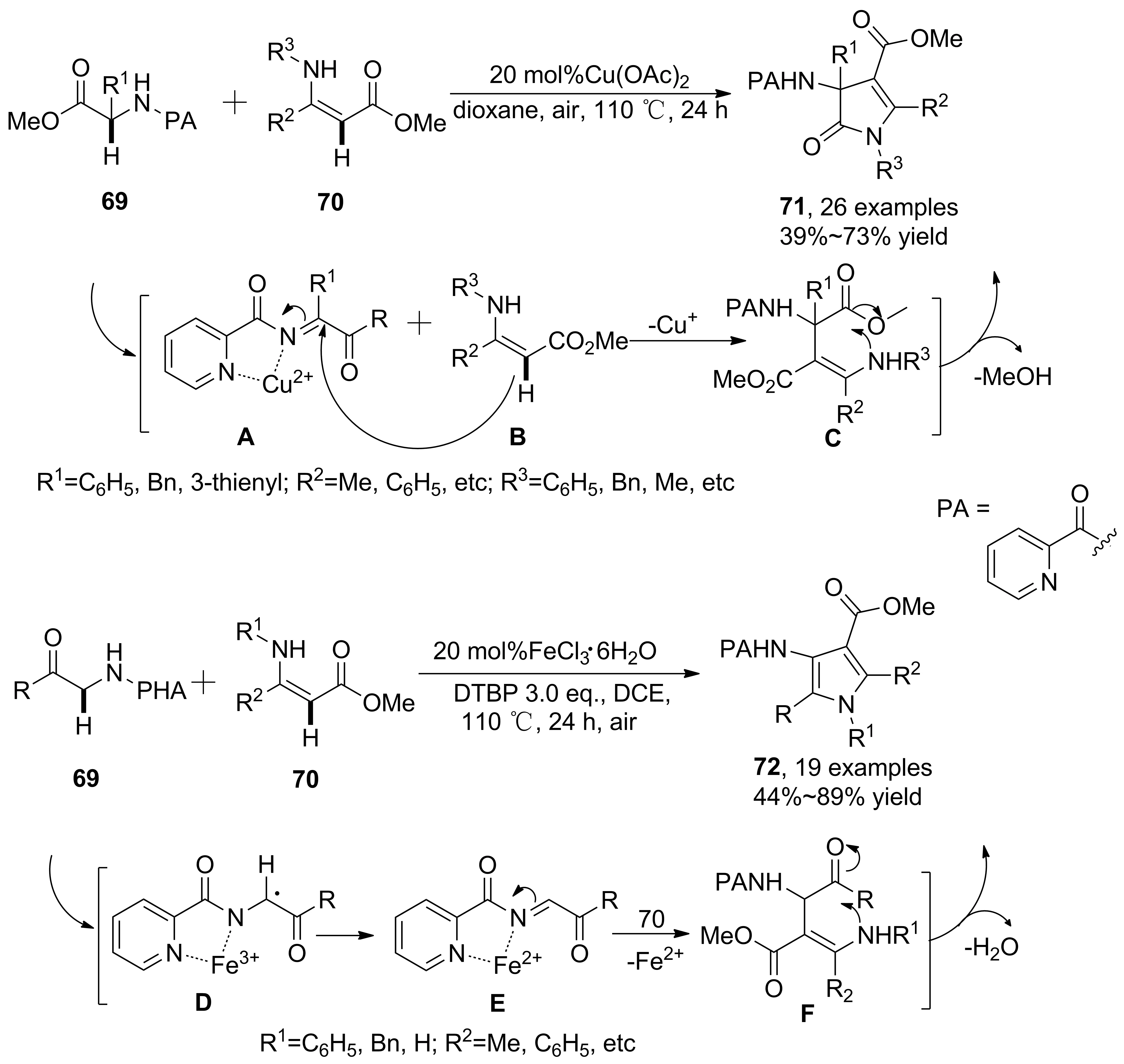
2016年,You等[43]報道了一種基于氨基酸酯、烯胺酯參與的兩組分串聯(lián)反應。采用二價銅離子催化,首次實現(xiàn)氨基酸酯羰基α-位C(sp3)—H鍵活化,并發(fā)生交叉偶聯(lián)、分子內(nèi)環(huán)化,得到多取代吡咯衍生物(71, Scheme 20),收率51%~86%。氨基酸酯結(jié)構(gòu)中引入2-羰基吡啶基團有助于捕獲催化劑,作為定位基團使得N鄰位C(sp3)—H鍵活化,具有良好的底物適用性,甚至采用聯(lián)二烯胺酯參與反應,也能得到相應產(chǎn)物。而在采用FeCl3·6H2O催化、DTBP作氧化劑時,則得到多取代3-氨基吡咯衍生物(72),收率55%~95%。其他Lewis酸,如AlCl3, ZnCl2和Zn(OTf)2均不能催化該反應,F(xiàn)eCl3·6H2O和DCE的組合顯示了反應特異性。機理研究表明,在FeCl3·6H2O催化體系中加入自由基捕獲劑(TEMPO)后,收率明顯下降,可能經(jīng)歷了自由基歷程。
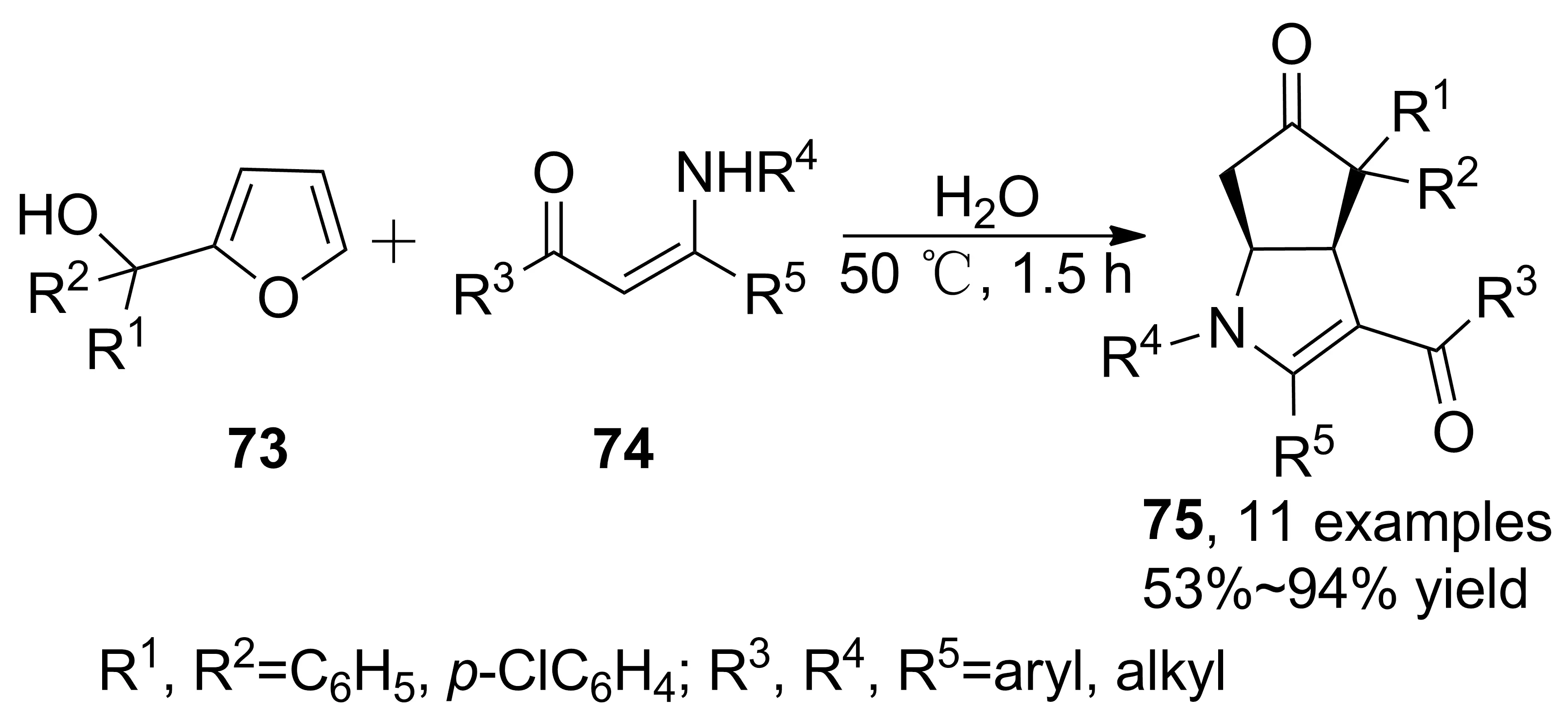
烯胺酮(酯)參與串聯(lián)反應合成吡咯衍生物在構(gòu)建復雜分子骨架方面也取得了較大發(fā)展。苯甲酰甲醛、靛紅、茚三酮、β-硝基烯烴等在該類串聯(lián)反應中被廣泛使用,近年來,其他種類烯胺酮(酯)參與的串聯(lián)反應也有所報道。2014年,Li等[44]道了ZnCl2催化烯胺酮與呋喃甲醇衍生物的串聯(lián)反應。首次利用烯胺酮C(2)-位點親核性進攻呋喃甲醇C(5)-位點,繼而發(fā)生Piancatelli重排、分子內(nèi)Michael加成,得到環(huán)戊二烯并[b]吡咯衍生物(75, Scheme 21),收率53%~94%。該反應解釋了Piancatelli重排親核碳進攻機理,反應對呋喃甲醇底物適用性有限,R1基團為正丁基或氫取代時反應不能發(fā)生。
2016年,Nguyen等[45]研究了2-氨基-1,4-萘醌、溴乙酰苯吡啶鹽、芳醛的串聯(lián)反應,實現(xiàn)一鍋法在萘醌上引入吡咯環(huán)(Scheme 22)。
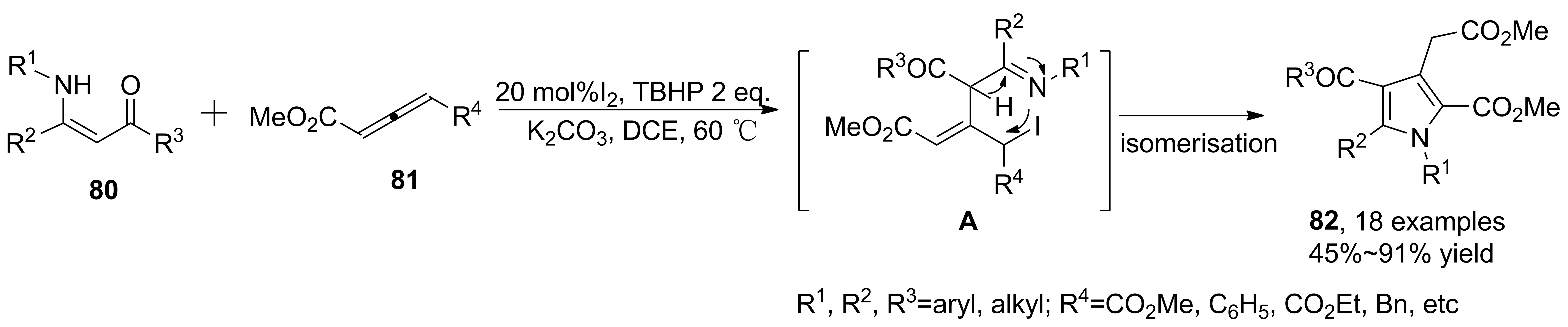
2016年,Deng等[46]報道了I2催化烯胺酯與聯(lián)烯酸酯的兩組分串聯(lián)反應,經(jīng)歷Michael加成、氧化環(huán)化得到多取代吡咯衍生物(82, Scheme23)。相比于之前的合成方法,只需催化量的I2即可完成轉(zhuǎn)化,可適用于不對稱取代的聯(lián)烯酸酯。
Scheme 18
Scheme 19
Scheme 20
Scheme 21

Scheme 22
Scheme 23
Scheme 24
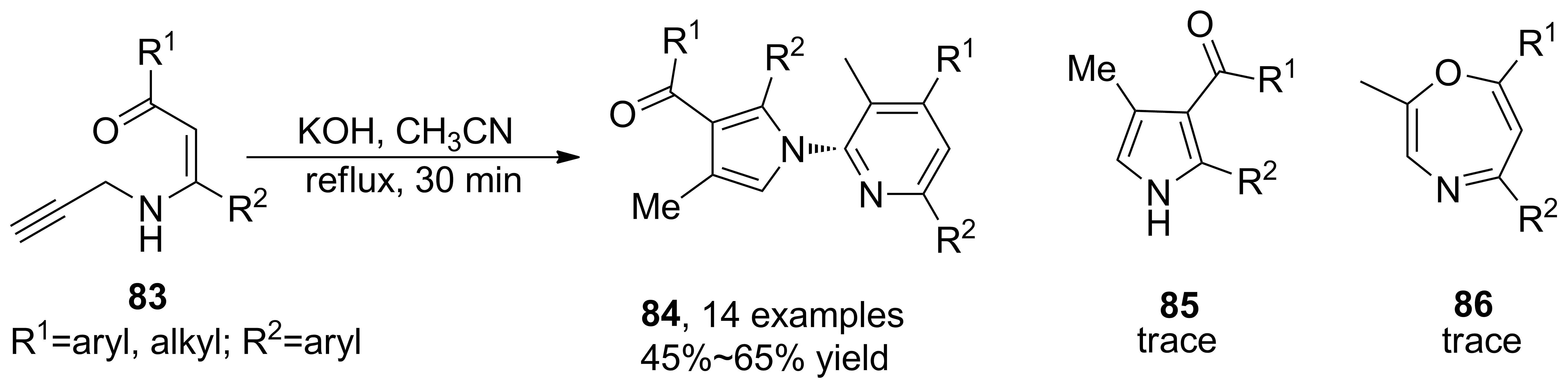
烯胺酮分子內(nèi)串聯(lián)環(huán)化方面也有所發(fā)展。2016年,Cui等[47]報道了KOH催化下烯胺酮分子內(nèi)環(huán)化構(gòu)建N-(2-吡啶基)吡咯衍生物的方法。與之前的方法需要事先加入吡啶不同,首次實現(xiàn)非金屬催化下同時構(gòu)建吡咯、吡啶環(huán)。KOH/CH3CN的組合實現(xiàn)產(chǎn)物的化學選擇合成,只得到化合物 (84, Scheme 24),收率28%~91%。烯胺酮底物結(jié)構(gòu)對產(chǎn)物影響較大,R1為叔丁基時反應失去選擇性,收率28%。若端炔上的氫替換成對甲氧基苯基或甲基時,只能得到吡咯環(huán)(85)。
烯胺酮(酯)結(jié)構(gòu)中富電子的烯胺與吸電子的羰基(酯基)相連,使得雙鍵進一步極化,作為多位點的有機合成子,參與的串聯(lián)反應在構(gòu)建雜環(huán)骨架方面已獲得長足的發(fā)展。苯甲酰甲醛、茚三酮、苊醌在吡咯稠環(huán)骨架修飾上被廣泛應用,實現(xiàn)了吲哚環(huán)、吡咯并吖啶環(huán)、吡咯并喹啉環(huán)、苊并[1,2-b]吲哚環(huán)等骨架的合成;烯胺酯在Ag或Au催化下實現(xiàn)了烯胺酯α-位C(sp2)—H鍵與苯乙炔β-位C(sp)—H鍵交叉偶聯(lián)/環(huán)化;β-硝基烯烴在綠色合成吡咯環(huán)上也有所發(fā)展,無論是合成方法還是分子結(jié)構(gòu)多樣性都得到了極大豐富。近年,烯胺酮(酯)作為關鍵中間體參與串聯(lián)反應合成吡咯衍生物方面也獲得了很大進展,如:丁炔二酸二酯類、丙炔酸酯類化合物與二級胺反應直接生成烯胺酯參與串聯(lián)反應[48],進一步豐富了烯胺酮(酯)的應用范圍。
但是,我們也應該看到目前該領域所面臨的挑戰(zhàn)。從反應形式上來看,烯胺酮親電性的羰基、C(sp2)-N(tert)位點以及親核性的α-碳位點參與的串聯(lián)反應已有大量報道,但對于C=C雙鍵[49]斷裂后兩個碳原子同時參與反應的研究相對較少;對于烯胺酮α-位或β-位上C—H鍵活化后與相應試劑交叉偶聯(lián)參與的串聯(lián)反應,目前也僅有幾例相關報道,有待進一步擴展。從分子結(jié)構(gòu)上來看,如何更加方便、有效的通過串聯(lián)反應在吡咯結(jié)構(gòu)上引入炔基、吡啶基等基團,實現(xiàn)吡咯結(jié)構(gòu)修飾,也值得化學工作者更深入的研究。
烯胺酮(酯)以其豐富的反應特性長久以來都是化學研究的熱點,相信在不久的將來,更加新穎、有效的應用烯胺酮(酯)構(gòu)建雜環(huán)衍生物方法會被逐漸發(fā)現(xiàn),并在有機合成領域發(fā)揮其潛在的作用。
[1] Nicolaou K C, Edmonds D J, Bulger P G. Cascade reactions in total synthesis[J].Angew Chem Int Ed,2006,45(43):7134-7186.
[2] Nicolaou K C, Chen J S. The art of total synthesis through cascade reactions[J].Chem Soc Rev,2009,38(11):2993-3009.
[3] Cheng D J, Ishihara Y, Tan B,etal. Organocatalytic asymmetric assembly reactions:Synthesis of spirooxindoles via organocascade strategies[J].ACS Catal,2014,4(3):743-762.
[4] Greenhill J V. Enaminones[J].Chem Soc Rev,1977,6(3):277-294.
[5] Elassara A Z, El-Khair A A. Recent developments in the chemistry of enaminones[J].Tetrahedron,2003,59(43):8463-8480.
[6] Vohra R K, Bruneau C, Renaud J L. Lewis acid-catalyzed sequential transformations:Straightforward preparation of functional dihydropyridines[J].Adv Synth Catal,2006,348(18):2571-2574.
[7] Kascheres C.The chemistry of enaminones,diazocarbonyls and small rings:Authors’ contribution[J].J Braz Chem Soc,2003,14(6):945-969.
[8] Negri G, Kascheres C, Kascheres A J. Recent development in preparation,reactivity,and biological activity of enaminoketones and enaminothiones and their utilization to prepare heterocyclic compounds[J].J Heterocycl Chem,2004,41(4):461-491.
[9] Stanovnik B, Svete J. Synthesis of heterocycles from alkyl 3-(dimethylamino)propenoates and related enaminones[J].Chem Rev,2004,104(5):2433-2480.
[10] 李明,郭維斯,文麗榮,等. 烯胺酮的合成及其在有機合成中的應用[J].有機化學,2006,26(9):1192-1207.
[11] 曹碩,景艷鋒,劉云云,等. 基于缺電子烯胺交叉氨化過程的多組分反應研究進展[J].有機化學,2014,34(5):876-885.
[12] Wan J P, Gao Y. Domino reactions based on combinatorial bond transformations in electron-deficient tertiary enamines[J].Chem Rec,2016,16(3):1164-1177.
[13] Wan J P, Jing Y F, Hu C F,etal. Metal-free synthesis of fully substituted pyridinesviaring construction based on the domino reactions of enaminones and aldehydes[J].J Org Chem,2016,81(15):6826-6831.
[14] Bredereck H, Effenberger F, Bostch H,etal. Syntheses in the heterocyclic series.v.reactions of vinylogous carboxamides to heterocycles[J].Chem Ber,1965,98(4):1081-1086.
[15] Zhao Y L, Zhang F F, Yao W J,etal. Base-promoted approach to highly functionalized conjugated dienes through enamine migration[J].Eur J Org Chem,2015,36:7984-7991.
[16] Abdel-Khalik M M, Elnagdi M H, Agamy S M. Studies with functionally substituted heteroaromatics:The chemistry ofN-phenylhydrazonylalkylpyridinium salts and of phenylhydrazonylalkylbenzoazoles[J].Synthe-sis,2000,2000(8):1166-1169.
[17] Liu J Y, Li Q Y, Jiang B,etal. Three-component domino reactions providing rapid and efficient routes to fully substituted pyrroles[J].RSC Advances,2013,3(3):5056-5068.
[18] Dhinakaran I, Padmini V, Bhuvanesh N. Chemodivergent,one-pot,multi-component synthesis of pyrroles and tetrahydropyridines under solvent- and catalyst-free conditions using the grinding method[J].ACS Comb Sci,2016,18(5):236-242.
[19] Jiang B, Yi M S, Shi F,etal. A multi-component domino reaction for the direct access to polyfunctionalized indoles via intermolecular allylic esterification and indolation[J].Chem Commun,2012,48(6):808-810.
[20] Jiang B, Yi M S, Tu M S,etal. Bronsted acid-promoted divergent reactions of enaminones:Efficient synthesis of fused pyrroles with different substitution patterns[J].Adv Synth Catal,2012,354(13):2504-2510.
[21] Jiang B, Li Y, Tu M S,etal. Allylic amination andN-arylation based domino reactions providing rapid three-component strategies to fused pyrroles with different substituted patterns[J].J Org Chem,2012,77(17):7497-7505.
[22] Fu L P, Shi Q Q, Shi Y,etal. Three-component domino reactions for regioselective formation of bis-indole derivatives[J].ACS Comb Sci,2013,15(2):135-140.
[23] Wang H Y, Shi D Q. Efficient synthesis of functionalized dihydro-1Hindol-4(5H)onesviaone-pot three-component reaction under catalyst-free conditions[J].ACS Comb Sci,2013,15(5):261-266.
[24] Zhu Q N, Zhang Y C, Xu M M,etal. Enantioselective construction of tetrahydroquinolin-5-one-based spirooxindole scaffoldviaan organocatalytic asymmetric multicomponent [3+3] cyclization[J].J Org Chem,2016,81(17):7898-7907.
[25] Wang H Y, Lin L L, Lin W,etal. An efficient synthesis of pyrrolo[2,3,4-kl]acridin-1-one derivatives catalyzed by Lproline[J].Org Lett,2012,14(17):4598-4601.
[26] Jiang B, Wang X, Xu H W,etal. Highly selective domino multicyclizations for forming polycyclic fused acridines and azaheterocyclic skeletons[J].Org Lett,2013,15(7):1540-1543.
[27] Hao W J, Wang J Q, Xu X P,etal. I2/O2promoted domino reactions of isatins or 3hydroxyindolin-2-one derivatives with enaminones[J].J Org Chem,2013,78(24):12362-12373.
[28] Yu F C, Yan S J, Hu L,etal. Cascade reaction of isatins with heterocyclic ketene aminals: synthesis of imidazopyrroloquinoline derivatives[J].Org Lett,2011,13(18):4782-4785.
[29] Yu F C, Zhou B, Xu H,etal. Three-component synthesis of functionalized pyrrolo[3,4-c]quinolin-1-ones by an unusual reductive cascade reaction[J].Tetrahedron,2015,71(7):1036-1044.
[30] Xu H, Zhou P, Zhou B,etal. Convenient one-step synthesis of pyrrolo[3,4-c]quinolin-1-onesviaTMSCl-catalyzed cascade reactions of isatins andβ-enamino ketones[J].RSC Adv,2016,6(77):73760-73768.
[31] Xu H, Zhou B, Zhou P,etal. Correction:Insights into the unexpected chemoselectivity in bronsted acid catalyzed cyclization of isatins with enaminones:Convenient synthesis of pyrrolo[3,4-c]quinolin-1-ones and spirooxindoles[J].Chem Commun,2016,52(51):9471-9472.
[32] Jiang B, Li Q Y, Tu S J,etal. Three-component domino reactions for selective formation of indeno[1,2b]indole derivatives[J].Org Lett,2012,14(20):5210-5213.
[33] Zhao F J, Sun M Y, Dang Y Y,etal. Domino bicyclization of 2,2-dihydroxyindene-1,3-dione with cyclic enaminones leading to isochromeno[4,3-b]indoles[J].Tetrahedron,201 4,70(51):9628-9634.
[34] Muthusaravanan S, Sasikumar C, Bala D B,etal. An eco-friendly three-component regio- and stereoselective synthesis of highly functionalized dihydroindeno[1,2-b]pyrroles under grinding[J].Green Chem,2014,16(3):1297-1304.
[35] Zhang J J, Feng X, Liu X C,etal. An efficient three component synthesis of highly functionalized tetrahydroacenaphtho[1,2-b]indolone derivatives catalyzed by L-proline[J].Mol Divers,2014,18(4):727-736.
[36] Chen X B, Luo T B, Guo G Z,etal. Selective synthesis of acenaphtho[1,2-b]indole derivatives via tandem regioselective aza-ene addition/N-cyclization/SN1 type reaction[J].Asian J Org Chem,2015,4(9):921-928.
[37] Fan W, Li Y R, Li Q,etal. Domino reactions of cyclic enaminones leading to selective synthesis of pentacyclic indoles and its functionalization[J].Tetrahedron,2016,72(32):4867-4877.
[38] Rueping M, Parra A. Fast,efficient,mild,and metal-free synthesis of pyrroles by domino reactions in water[J].Org Lett,2010,12(22):5281-5283.
[39] Guan Z H, Li L, Ren Z H,etal. A facile and efficient synthesis of multisubstituted pyrroles from enaminoesters and nitroolefins[J].Green Chem,2011,13(7):1664-1668.
[40] Zhao M N, Liang H, Ren Z H,etal. Iron-catalyzed tandem one pot addition and cyclization of the Blaise reaction intermediate and nitroolefins. synthesis of substituted NH-pyrroles from nitriles[J].Adv Synth Catal,2013,355(1):221-226.
[41] Ke J, He C, Liu H Y,etal. Oxidative cross-coupling/cyclization to build polysubstituted pyrroles from terminal alkynes andb-enamino esters[J].Chem Commun,2013,49(68):7549-7551.
[42] Zhang S, Ma Y,Lan J B,etal. Gold catalyzed cascade C—H/C—H cross-coupling/cyclization/alkynylation:An efficient access to 3-alkynylpyrroles[J].Org Biomol Chem,2015,13(21):5867-5870.
[43] Li K Z, You J S. Cascade oxidative coupling cyclization:A gateway to 3-amino polysubstituted five-membered heterocycles[J].J Org Chem, 2016,81(6):2327-2339.
[44] Wang C Y, Dong C Y, Kong L K,etal. ZnCl2catalyzed chemoselective cascade reactions of enaminones with 2-furylcarbinols:A versatile process for the synthesis of cyclopenta[b]pyrrole derivatives[J].Chem Commun,2014,50(17):2164-2166.
[45] Nguyen T Q, Nhat T G L, Ngoc D V,etal. Synthesis of novel 2-aryl-3-benzoyl-1H-benzo[f]indole-4,9-diones using a domino reaction[J].Tetrahedron Lett.2016,57(39):4352-4355.
[46] Wang Y, Jiang C M, Li H L,etal. Regioselective iodine-catalyzed construction of polysubstituted pyrroles from allenes and enamines[J].J Org Chem,2016,81(18):8653-8658.
[47] Shen J H, Yang X F, Wang F Y,etal. Base-mediated regiospecific cascade synthesis ofN-(2-pyridyl)pyrroles fromN-propargylicβ-enaminones[J].RSC Adv,2016,6(54):48905-48909.
[48] Wang H Y, Liu X C, Feng X,etal. GAP chemistry for pyrrolyl coumarin derivatives:A highly efficient one-pot synthesis under catalyst free conditions[J].Green Chem,2013,15(12):3307-3311.
[49] Wan J P, Zhou Y Y, Cao S. Domino reactions involving the branched C—N and C=C cleavage of enaminones toward pyridines synthesis[J].J Org Chem,2014,79(20):9872-9877.
《合成化學》致謝審稿專家
在過去的半年中,以下專家學者為《合成化學》的學術(shù)質(zhì)量把控提供了重要幫助(排名不分先后):
鄧志勇 張曉梅 彭 林 陳永正 徐小英 王啟衛(wèi) 白 威 嚴思明 張 瑞 于曉榮
李小可 焦利賓 劉 舉 鄭玉國 鄭欣梅 周 英 劉雄利 尹志偉 周曉靚 吳 琴
張 昭 金 燦 張 敏 吳杰穎 劉 霞 楊建新 劉新華 張淑華 焦 銳 錢 廣
徐 峰 薛 偉 李邦玉 伍 勇 王平保 吳永平 王健春 胡 育 曹志凌 葛洪玉
李春霞 徐 亮 何 菱 黃青春 李公春 黃焰根 盧久富 吳麗穎 關 磊 張 建
敖桂珍 王 京 毛澤偉 張 磊 楊 鵬 汪鵬飛 楊 鵬 史大斌 崔漢峰 蔡志強
左勝利 黃 雁 丁 瑜 王 鋒 白躍飛 徐 鑒 路德待 宋冰蕾 張東峰 毛武濤
佘世雄 涂國剛 楊雪梅 楊 銘 吳 燕 樂傳俊 劉進兵 魏夢雪 宋 顥 尹曉剛
寇玉輝 黃統(tǒng)輝 鐘 錚 袁偉成 王治明 卓廣瀾 江健安 高復興 王祖利 段群鵬
王 凱 郭其祥 張來軍 劉 杰 薛志勇 倪士峰 鄭廣兵 楊尊華 劉 波 禹興海
梁興華 袁澤利 蔡志彬 王曉玲 梁作芹
特此致謝!
《合成化學》編輯部
Research Progress in The Synthesis of Pyrrole Derivatives by Domino Reaction of Enamine(Enamioesters)
FU Hong-liang, WANG Hui, WU Yi-bing, LING Fei, ZHONG Wei-hui*
(Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals,College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, China)
As multi-site organic synthons, enamines or enamioesters have been broadly employed in the synthesis of pyrrole derivativesviadomino reaction. The research progress of enamines(enamioesters) in the synthesis of pyrrole monoheterocycles and fused heterocycles through domino reaction withortho-dicarbonyl compounds,β-nitroalkenes or phenylacetylenes was reviewed with 49 references. The outlook was briefly described as well.
enamine(enamioester); domino reaction; pyrrole derivative; research progress; review
2017-02-14; 修改日期: 2017-06-20
國家自然科學基金資助項目(21676253)
傅鴻樑(1991-) ,男,漢族,浙江紹興人,碩士研究生,主要從事手性藥物及中間體的合成研究。 E-mail: elijah7119@163.com
鐘為慧,教授,博士生導師, E-mail: weihuizhong@zjut.edu.cn
O626.13
A
10.15952/j.cnki.cjsc.1005-1511.2017.08.17024
