顱內間葉型軟骨肉瘤1例并文獻回顧
2017-08-21 08:55:26顧昕馮睿
復旦學報(醫學版) 2017年4期
顧 昕 馮 睿
(1上海市靜安區中心醫院神經外科 上海 200040; 2復旦大學附屬華山醫院神經外科 上海 200040)
顱內間葉型軟骨肉瘤1例并文獻回顧
顧 昕1△馮 睿2
(1上海市靜安區中心醫院神經外科 上海 200040;2復旦大學附屬華山醫院神經外科 上海 200040)
Mott[1]于1899年報道了第1例顱內軟骨肉瘤(intracranial condrosarcoma,IC)。隨著學者們對腫瘤不同分型的重視,Lichtenstein等[2]于1959年首先報道了間葉型軟骨肉瘤(mesenchymal condrosarcoma,MC),描述其為一種惡性程度高的、侵襲性強的腫瘤。Dahlin等[3]在1962最先報道了1例顱內間葉型軟骨肉瘤(intracranial mesenchymal condrosarcoma,IMC)。IC只占所有原發顱內占位的0.15%[4],十分罕見,大部位于顱底,IMC更只占其中的30%[5],目前英文文獻報道約40例。IMC在臨床特征上有異于其他IC的獨特特征,特此討論。
臨床資料 患者女性,41歲,因“左下肢乏力伴行走不穩2個月”入院。2個月前無明顯誘因首發左拇趾麻木、無力,逐漸向上發展至左下肢無力,行走困難需攙扶。左上肢乏力不明顯。右側肢體無影響。
查體 神志清楚,顱神經檢查未見明顯異常,左下肢肌力髖、膝、踝關節均為Ⅱ級,趾關節Ⅱ-級,左上肢肌力肩、肘、腕、指關節均為Ⅴ-級,右側肢體肌力Ⅴ級;左下肢肌張力呈折刀樣增高,左上肢及右側無明顯異常;左側肢體膝反射、跟腱反射增強(+++),余肢體腱反射正常;左側巴氏征、奧本海默征等病理征可疑陽性(+/-)。
影像學 頭顱MRI平掃(圖1)示占位大小約5 cm×6 cm×4 cm,主體位于右側額葉后部及大腦鐮旁,少量延伸至左側,周圍環形水腫帶明顯,T1、T2加權呈等、低混雜信號,增強后邊界清楚,呈明顯、欠均勻的強化,并可見腦膜尾征,中央可見低信號片狀影,囊變亦可見,中線稍向左側移位約2 mm。MRV示額部上矢狀竇顯示不清,周圍可見走行扭曲紊亂的小血管影。術前診斷為“雙側額后、鐮旁腦膜瘤”。
手術治療 完善術前檢查后全麻下行開顱腫瘤切除術,取額后冠狀切口,右側骨瓣銑開,切開右額硬腦膜即見腫瘤位于矢狀竇旁,小心止血、分離腫瘤邊界后,鏟除與矢狀竇、大腦鐮粘連的腫瘤基底部,然后分塊切除腫瘤,腫瘤質地偏硬,血供豐富,出血較多;腫瘤部分長入矢狀竇內,粘連緊密,難以完全切除,最終達次全切除。術中輸血200 mL×2次。術后當日患者左下肢肌力恢復至髖、膝、踝、趾Ⅲ級,3日后在家屬攙扶下開始下地行走,病程中患者情況平穩無特殊,術后11天出院時肌力檢查:髖、膝、踝、趾均Ⅳ+級,右下肢腱反射增強。術后病理結果為:IMC。經充分溝通后,患者及家屬決定回當地進一步治療,術后1個月左右患者接受普通放療+靜脈化療(藥物不詳)。患者術后3個月復查MRI增強掃描(圖2),顯示病灶處無明顯復發征象,瘤腔清晰可見,矢狀竇內少量腫瘤殘留可能。
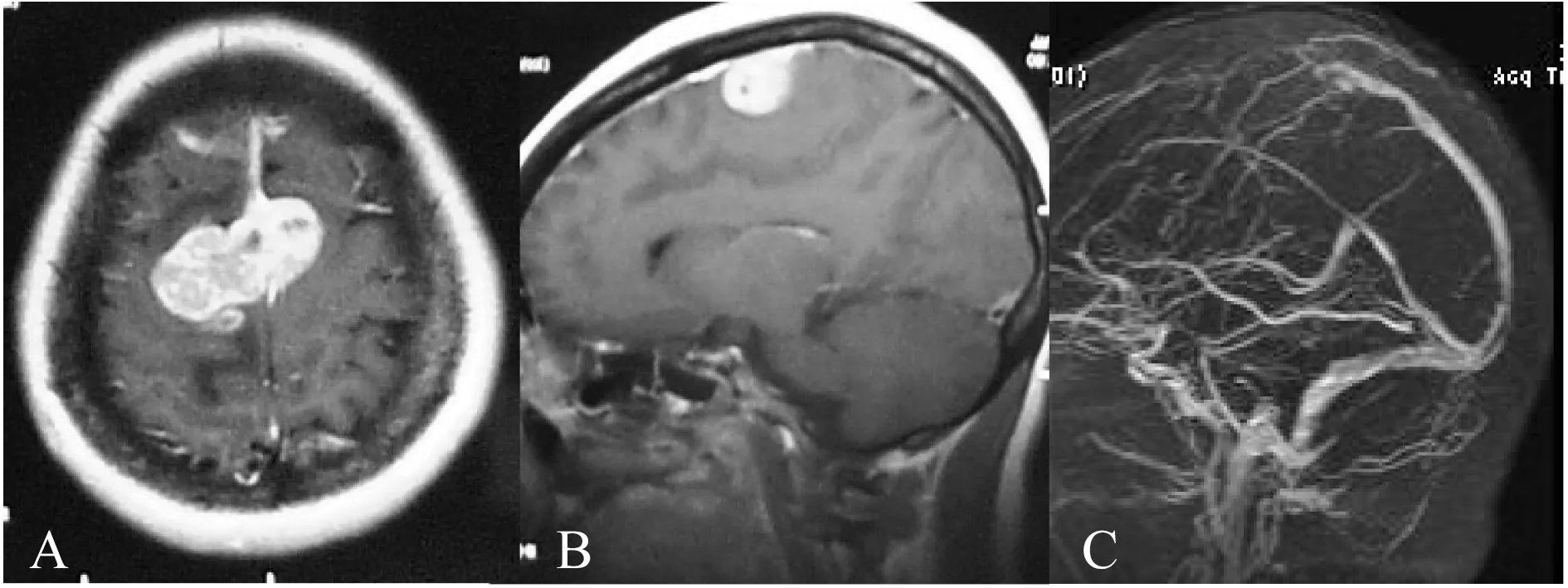
A:Preoperative axial;B:Sagittal enhanced MRI showed bilateral parasagittal tumors;C:MRV showed no patency of the sagittal sinus.
圖1 術前增強MRI和MRV
Fig 1 Enhanced MRI and MRV findings before operation

A:3 months after operation,the MRI level showed a small residual in the sagittal sinus; B:No obvious sign of recurrence in sagittal plane.
圖2 術后3個月增強MRI
Fig 2 Enhanced MRI findings 3 months after operation
病理特點 大體標本呈分葉狀,可見出血、鈣化及囊變,切面灰白質韌。鏡下見(圖3):梭形瘤細胞片巢狀或編織狀密集排列,核染色深,核分裂相可見,小血管豐富,部分區域可見軟骨形成、骨化、鈣化,分化好的軟骨區域與未分化的間質交錯分布,二者之間的交界緣清晰。免疫組化:Vim(+),CD99(+),CD34(-),PR(-),EMA(-/+),S100(+),D2-40(-/+),MIB-1:40%。

A:A dense arrangement of cells in a nest or braid (HE);B:The positive expression of S100 tumor nuclei.
圖3 腫瘤病理鏡下表現(×100)
Fig 3 Pathological appearance of tumor (×100)
討論 IMC多發于10~30歲的年輕人群[6-7]。Chen等[8]回顧的37例患者平均年齡約21.5歲,男女比例為16∶21。間葉型有女性多發傾向[9-10],黏液型在人群統計學上與間葉型類似[11],而傳統型無明顯年齡、性別傾向。顱內是骨外MC最常發的部位[12-13]。復旦大學附屬華山醫院于2008年曾報道7例IC,其中4例是IMC,均位于顱底[14]。Korten等[5]于1998年回顧了192例IC,統計結果表明:其中超過80%在顱底部位。但IMC的實際多發位置卻在非顱底處。2004年Chen等[8]回顧了37例IMC患者,28/37例位于非顱底處,且位置傾向于出現在額頂區域,右側更多[6,12]。值得一提的是,絕大部分位于大腦鐮及矢狀竇旁的IC都是間葉型,其他類型在此處極為罕見[11,15]。關于IMC的起源問題,目前沒有定論:顱底處的IMC可認為起源于顱底軟骨聯合處的未分化胚胎細胞[3,11],但大部分IMC位于非顱底處。目前較流行的學說認為IMC源于硬膜或蛛網膜中原始的多能間質細胞,它們有生成軟骨肉瘤的潛能[11]。也有人認為IMC起源于腦膜纖維母細胞或分化中的前間質軟骨祖細胞[16]。
根據WHO對骨腫瘤的分類,軟骨肉瘤病理分型為[17]:傳統型,間葉型,透明細胞型和去分化型。而IC有特別的分類,Russell等[18]根據組織學結構已將IC分為傳統型、間葉型和黏液型(中間型),這種分類一直延續至今,間葉型和黏液型分開歸類的提出是為了區別于傳統型的Ⅰ~Ⅲ分級[5],間葉型分級本身意味著侵襲性強、轉歸差。Lichtenstein等[2]最早建議MC為一種單獨的亞型,Salvati等[13]以及Korten等[5]也認為間葉型應該被歸為單獨的一類。目前尚未在顱內發現透明細胞型或去分化型軟骨肉瘤[17]。即IC的分型僅有傳統型和間葉型兩類。
傳統型軟骨肉瘤可有典型的蜂窩狀增強[15],而IMC無此特點。CT或平片常可見占位內的點片狀鈣化[19],T2WI常可見鈣化區+非鈣化區2個區域,增強MRI可見不均勻強化[20]。超過50%的腫瘤在影像學上有顯著的富血管性,高度疑似腦膜瘤或血管外皮瘤,有些甚至可與動靜脈畸形混淆。然而,這些影像學特征都缺乏敏感性和特異性,因此影像學診斷十分困難。
IMC現今首選的治療方案仍然是外科切除,繼而輔以放化療。外科全切除腫瘤已是公認的最佳和首要治療方案[13,21],切除程度是決定長期預后的關鍵,因全切除后患者常可獲得較長生存期。真正意義上的腫瘤全切除邊界應超過腫瘤的實際邊界(即擴大切除),因為研究表明MC會有偽足樣突起突入周圍軟組織[22]。生長在顱底、侵犯顱內重要靜脈竇以及術前誤診為良性轉歸的腦膜瘤的病例,往往得不到真正的全切除,因此后續治療十分必要,即便全切除的病例也應積極行后續治療。
放射治療應用十分廣泛,已成為現今MC外科治療后首選的后續治療方案[22]。在大多數IMC病例放療被證明能夠減輕影像學增強程度和減小占位體積[23-24]。后續進行化療的病例極少,且大部分學者認為化療對IMC無效。然而Spina等[7]報道的1例IMC,術后應用類肉瘤方案進行化療,療程結束后增強MRI顯示病灶處強化明顯減弱。Aksoy等[25]用替莫唑胺治療復發的顳頂葉IMC,患者癥狀幾乎完全消失,MRI也顯示病灶體積明顯減小。
IMC的轉歸常為短期內的原位復發或遠處轉移[26]。傳統型與黏液型IC僅有6%的5年死亡率,而IMC的5年死亡率高達54%[17]。傳統型和黏液型IC的預后取決于腫瘤的分級(Ⅰ~Ⅲ級),而間葉型本身就意味著腫瘤級別高、侵襲型強,預后極差[4]。
總之,IMC是一種罕見的、高度惡性的腫瘤,它在IC中惡性程度最高、轉歸最差,多發于非顱底的位置,常位于額頂葉處,多見于10~30歲的年輕人;位于鐮旁者絕大多數為IMC。IMC病理學特點為軟骨區域與間質區域的雙相性分布。影像學表現缺乏特異性和敏感性,術前往往誤診為腦膜瘤。外科擴大切除是最佳和首選的治療方案,但切除后往往復發快,有時可遠處轉移,必須行后續治療。放射治療是首選的、被普遍接受的后續治療;大部分學者對化療持保留意見,而且接受此治療的病例極少,但已有人報道了成功化療的病例,這也許是IMC治療新的突破口。在臨床工作中,對位于非顱底部位特別是矢旁或鐮旁的酷似腦膜瘤的占位,如同時伴有上述特征,應將IMC列為鑒別診斷之一。
顱內間葉型軟骨肉瘤; 外科治療; 放射治療;化學治療
[1] MOTT FW.Chondro-sarcoma springing from the sella turcica[J].ArchNeurolPsychiat,1899,1:432-433.
[2] LICHTENSTEIN L,BERNSTEIN D.Unusual benign and malignant chondroid tumors of bone- a survey of some mesenchymal cartilage tumors and malignant chondroblastic tumors,including a few multicentric ones,as well as many atypical benign chondroblastomas and chondromyxoid fibromas[J].Cancer,1959,12(6):1142-1157.
[3] DAHLIN DC,HENDERSON ED.Mesenchymal chondrosarcoma.Further observations on a new entity[J].Cancer,1962,15(2):410-417.
[4] CHANDLER JP,YASHAR P,LASKIN WB,etal.Intracranial chondrosarcoma:a case report and review of the literature[J].JNeuro-Oncol,2004,68(1):33-39.
[5] KORTEN A,TER BERG HJW,SPINCEMAILLE GH,etal.Intracranial chondrosarcoma:review of the literature and report of 15 cases[J].JNeurolNeurosurPS,1998,65(1):88-92.
[6] CROSSWELL H,BUCHINO JJ,SWEETMAN R,etal.Intracranial mesenchymal chondrosarcoma in an infant[J].MedPediatrOncol,2000,34(5):370-374.
[7] LA SPINA M,DOLLO C,GIANGASPERO F,etal.Intracranial mesenchymal chondrosarcoma with osteoid formation:report of a pediatric case[J].ChildNervSyst,2003,19(9):680-682.
[8] CHEN JY,HSU SS,HO JT.Extraskeletal intracranial mesenchymal chondrosarcoma:case report and literature review[J].KaohsiungJMedSci,2004,20(5):240-246.
[9] BINGAMAN KD,ALLEYNE CH,OLSON JJ.Intracranial extraskeletal mesenchymal chondrosarcoma:Case report[J].Neurosurg,2000,46(1):207-211.
[10] CHEN JY,HSU SS,HO JT.Extraskeletal intracranial mesenchymal chondrosarcoma:case report and literature review[J].KaohsiungJMedSci,2004,20(5):240-246.
[11] ORUCKAPTAN HH,BERKER M,SOYLEMEZOGLU F,etal.Parafalcine chondrosarcoma:An unusual localization for a classical variant-case report and review of the literature[J].SurgNeurol,2001,55(3):174-179.
[12] DE CECIO R,MIGLIACCIO I,FALLETI J,etal.Congenital intracranial mesenchymal chondrosarcoma:Case report and review of the literature in pediatric patients[J].PediatrDevelPathol,2008,11(4):309-313.
[13] SALVATI M,CAROLI E,FRATI A,etal.Central nervous system mesenchymal chondrosarcoma[J].JExpClinCancRes,2005,24(2):317-324.
[14] 張榮.顱底軟骨肉瘤的診治與預后(7例報告)[J].中國神經精神疾病雜志,2008,34(4):229-232.
[15] KATHIRAVEL Y,FINNIS NDM.Primary falcine chondrosarcoma[J].JClinNeurosci,2008,15(12):1406-1409.
[16] AIGNER T,LOOS S,MULLER S,etal.Cell differentiation and matrix gene expression in mesenchymal chondrosarcomas[J].AmJPathol,2000,156(4):1327-1335.
[17] BLOCH OG,JIAN BJ,YANG I,etal.A systematic review of intracranial chondrosarcoma and survival[J].JClinNeurosci,2009,16(12):1547-1551.
[18] RUSSELL DS.Meningeal tumours:a review[J].JClinPathol,1950,3(3):191-211.
[19] RUSHING EJ,ARMONDA RA,ANSARI Q,etal.Mesenchymal chondrosarcoma-A clinicopathologic and flow cytometric study of 13 cases presenting in the central nervous system[J].Cancer,1996,77(9):1884-1891.
[20] HASHIMOTO N,UEDA T,JOYAMA S,etal.Extraskeletal mesenchymal chondrosarcoma:an imaging review of ten new patients[J].SkeletalRadiol,2005,34(12):785-792.
[21] BOSE B.Intracranial extraskeletal mesenchymal chondrosarcoma:Case report and review of the literature[J].NeurosurgQuart,2003,13(1):30-39.
[22] PELLITTERI PK,FERLITO A,FAGAN JJ,etal.Mesenchymal chondrosarcoma of the head and neck[J].OralOncol,2007,43(10):970-975.
[23] HUG EB,LOREDO LN,SLATER JD,etal.Proton radiation therapy for chordomas and chondrosarcomas of the skull base[J].JNeurosurg,1999,91(3):432-439.
[24] HOSHINO M,TANJI H,WATANABE M,etal.A case of intracranial mesenchymal chondrosarcoma-changes observed by computed tomography before and after radiotherapy (author′s transl)[J].NoShinkeiGeka,1981,9(7):843-848.
[25] AKSOY S,ABALI H,KILICKAP S,etal.Successful treatment of a chemoresistant tumor with temozolomide in an adult patient:report of a recurrent intracranial mesenchymal chondrosarcoma[J].JNeuro-Oncol,2005,71(3):333-334.
[26] SCHEITHAUER BW,RUBINSTEIN LJ.Meningeal mesenchymal chondrosarcoma-report of 8 cases with review of the literature[J].Cancer,1978,42(6):2744-2752.
R739.41
B
10.3969/j.issn.1672-8467.2017.04.030
2016-08-11;編輯:王蔚)
△Corresponding author E-mail:guxin93@sina.com
