抗腫瘤多肽9R-P201誘導下的肝癌HepG2細胞轉錄組測序分析
2017-07-12 18:05:57劉文榮丁若凡張一鳴李宇鵬李玲郭志云
生物技術通報 2017年7期
劉文榮丁若凡張一鳴李宇鵬李玲郭志云
(1. 西南交通大學生命科學與工程學院,成都 610031;2. 成都市第三人民醫院病理科,成都 610031)
抗腫瘤多肽9R-P201誘導下的肝癌HepG2細胞轉錄組測序分析
劉文榮1丁若凡1張一鳴1李宇鵬1李玲2郭志云1
(1. 西南交通大學生命科學與工程學院,成都 610031;2. 成都市第三人民醫院病理科,成都 610031)
旨在探索多肽9R-P201處理肝癌HepG2細胞后基因融合、單核苷酸多態性(Single nucleotide polymorphism,SNP)突變、可變剪接等事件,并分析差異表達基因所參與的生物學進程與信號通路,以期解析多肽9R-P201在轉錄組水平對肝癌細胞的調控。通過轉錄組測序檢測9R-P201處理肝癌HepG2細胞前后基因差異表達情況,tophat-fusion軟件檢測基因融合,SAMTOOLS軟件檢測SNP位點,rMATS軟件鑒定可變剪接,使用基因本體(Gene Ontology,GO)和京都基因與基因組百科全書(Kyoto encyclopedia of genes and genomes,KEGG)富集分析方法對差異表達基因進行功能富集分析。結果共檢測到可變剪接事件276個、SNP位點5 557個、基因融合事件45個;同時共得到顯著差異表達基因 403個,其中上調269個而下調134個,基因的功能富集分析結果顯示差異表達基因顯著富集細胞生長、遷移等腫瘤相關生物進程,并參與多條與癌癥相關的信號通路。研究表明在9R-P201誘導HepG2細胞后,導致表達差異基因顯著與腫瘤生物學進程和通路相關,并發生了大量可變剪接、SNP突變、基因融合等事件,這暗示著該多肽有望作為后續肝癌介入治療潛在藥物分子。
9R-P201;肝細胞癌;轉錄組測序;基因
在全世界范圍內,肝細胞癌(Hepatocellular carcinoma,HCC)是最為常見的惡性腫瘤之一,死亡率高居所有腫瘤第二位[1]。目前除了肝移植、手術以及放化療等治療手段外,分子靶向藥物在治療肝細胞癌方面也取得了許多重要的進展[2]。目前靶向肝細胞癌藥物主要包括化學藥物和多肽藥物,其中索拉菲尼、舒尼替尼、瓦他拉尼等分子靶向化學藥物已在臨床上應用[3]。除此之外,靶向肝細胞癌的多肽藥物的研究也取得明顯進展,多肽藥物是由人工合成的或經分離純化得到的活性多肽,先前研究發現一些小分子多肽在抑制腫瘤的發生與發展中起著重要的作用。例如,蛋氨酸腦啡肽(Methionine enkephalin,MENK)是具有增強體液免疫和提高細胞殺傷作用的內源性多肽,研究表明MENK通過精確調控樹狀細胞的阿片類藥物受體介導的功能來發揮抗腫瘤的活性[4]。研究證實從家蠶幼蟲中分離得到的抗菌肽CecropinXJ 能抑制肝癌細胞的生長并誘導凋亡,這暗示著CecropinXJ 或許可作為抗肝癌的多肽藥物[5]。
本實驗室前期通過噬菌體隨機12肽庫篩選得到了一個多肽9R-P201,實驗證實9R-P201可顯著抑制肝癌HepG2細胞的存活、增殖和遷移并誘導凋亡[6,7]。為了探尋多肽9R-P201在轉錄組水平抑制肝細胞癌的形成發展的機理,本研究利用轉錄組測序對9R-P201處理HepG2細胞前后的基因表達譜進行分析,篩選出了一系列差異表達基因,并證明其生物學功能。
1 材料與方法
1.1 材料
人肝癌細胞HepG2細胞株來自于四川大學生命學院,由本實驗室保管;9R-P201購自上海強耀生物科技有限公司;DMEM培養基購自Gibco公司;100 U/mL青霉素和100 U/mL鏈霉素購自Hyclone公司;胎牛血清購自Biowest公司;Trizol Regent購自Invitrogen公司;轉錄組測序由北京百邁客生物科技有限公司完成。
1.2 方法
1.2.1 細胞培養 HepG2細胞用含有10% 胎牛血清(Biowest),100 IU/mL青霉素和100 mg/mL鏈霉素(Hyclone)的DMEM培養基(Gibco),并置于37℃、5% CO2飽和濕度的細胞培養箱中進行培養。取處于對數生長期的細胞3.0×105個接種于6孔板中,培養過夜至細胞融合度達到80 %左右,用80 μg/mL的9R-P201(上海強耀生物)作用于HepG2細胞,處理24 h后,收集細胞用于后續的RNA提取。
1.2.2 RNA提取及質量檢測 收集好的9R-P201處理和未處理的HepG2細胞用Trizol試劑(Invitrogen)按試劑說明書提取細胞總RNA。1 %瓊脂糖凝膠電泳和酶標儀(Biotech)鑒定RNA的純度、濃度及完整性,合格的總RNA保存于-80℃冰箱待用。
1.2.3 RNA文庫構建、測序 RNA檢測合格后,用帶有Oligo(dT)的磁珠進行富集并隨機打斷,以短片段RNA為模板,用六堿基隨機引物(random hexamers)合成一鏈cDNA,然后加入緩沖液、dNTPs、RNase和DNA polymerase I合成二鏈cDNA。純化的雙鏈cDNA再進行末端修復、加A尾并連接測序接頭,最后進行PCR富集得到鏈特異性cDNA文庫,使用Illumina HiSeq 4000測序儀進行文庫測序。
1.2.4 轉錄組測序數據的處理、比對和組裝 測序原始測序數據通過去除帶接頭和低質量的reads后獲得高質量的clean reads。使用Tophat2軟件將clean reads比對到hg19參考基因組上,用Cufflinks軟件按照hg19參考基因組進行組裝[8]。
1.2.5 可變剪接鑒定、基因融合事件鑒定和SNP位點分析 使用rMATS軟件進行可變剪接的鑒定,在本次轉錄組測序分析中我們主要對常見的5種可變剪接類型進行統計。使用tophat-fusion軟件來鑒定基因融合,最后使用Circos可視化工具來繪制基因融合示意圖[9]。將每個樣品的測序結果與參考基因組相比,使用SAMTOOLS軟件檢測SNP位點,并使用ANNOVAR工具對SNP位點進行注釋[10]。
1.2.6 差異表達基因及功能富集分析 采用Audic S算法進行差異表達分析,將錯誤發現率(False discovery rate,FDR)校正后的q< 0.05且差異倍數|log2FC|≥1的視為差異表達基因,并對篩選得到的差異表達基因進行聚類分析。利用GO數據庫和KEGG數據庫對差異表達的基因進行GO與信號通路富集分析,以q < 0.05為顯著性閾值[11,12]。
2 結果
2.1 轉錄組測序與差異分析基因分析
轉錄組測序得到對照組和處理組總的reads數分別有46 698 580和39 332 045條,過濾得到clean reads數所占的比例分別為99.45%和97.46%。本次轉錄組測序共檢測到18 152個基因,按照差異表達篩選標準得到差異表達基因 403個,其中上調269個下調134個,我們對篩選得到的差異表達基因分析表明9R-P201處理后基因整體呈上調趨勢(圖1)。
2.2 可變剪接與基因融合事件鑒定
對常見的5種可變剪接類型的數量進行了統計,結果發現在本次轉錄組測序中一共檢測到276個可變剪接,其中外顯子跳躍(Exon skipping,ES)最多有126個而內含子保留(Intron Retention,RI)最少有12個,此外3'端可變剪接(Alternative 3' splice site,A3SS)有54個、5'端可變剪接(Alternative 5' splice site,A5SS)有60個、互斥外顯子(Mutually exclusive exon,MXE)有24個(圖2-A)。
異常的融合基因可以引起惡性血液疾病以及腫瘤,為此進行了基因融合分析。結果共鑒定得到45個基因融合,發現基因融合事件主要發生在11號、12號和19號染色體上(圖2-B)。在這45個基因融合中,FTH1、GAPDH、KRT7等基因與其它基因發生融合的頻率比較高(表1)。
2.3 SNP分析
通過在對照組和處理組進行SNP分析發現在對照組和處理組中分別得到SNP位點2 180個和3 377個,其中對照組的2 180個SNP包括816個非同義突變SNP(Nonsynonymous SNP)和1 364個同義突變SNP(Synonymous SNP),處理組的3 377個SNP包括1 249個非同義突變SNP(Nonsynonymous SNP)和2 128個同義突變SNP(Synonymous SNP);同義突變SNP和非同義突變SNP中都包括6種類型SNP突變,結果(圖2-C)發現C/T的突變頻率最高而A/T的突變頻率最低。
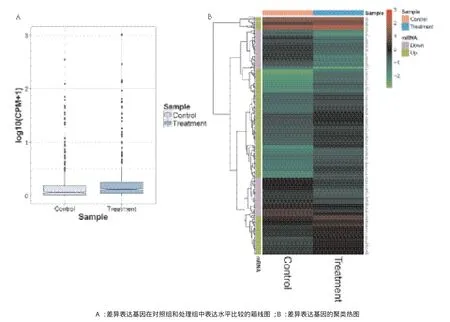
圖1 差異表達基因分析
2.4 差異表達基因功能分析
通過GO和KEGG富集分析對403個差異表達基因進行功能注釋,以q < 0.05為顯著性閾值。GO富集分析一共富集上34條GO term,包括生物學過程(BP)18條、細胞成分(CC)8條以及分子功能(MF)8條,其中富集上與腫瘤相關的GO term有細胞生長(q = 5.05E-08)、細胞移動(q = 8.19 E-07)、細胞信號轉導(q = 9.91 E-06)等(圖3-A)。KEGG信號通路富集分析結果(圖3-B)表明,差異表達基因顯著地富集上腫瘤轉錄失調(q = 4.66 E-08)、細胞因子受體相互作用(q = 3.84 E-06)、細胞間隙連接(q = 3.40E-05)等信號通路。
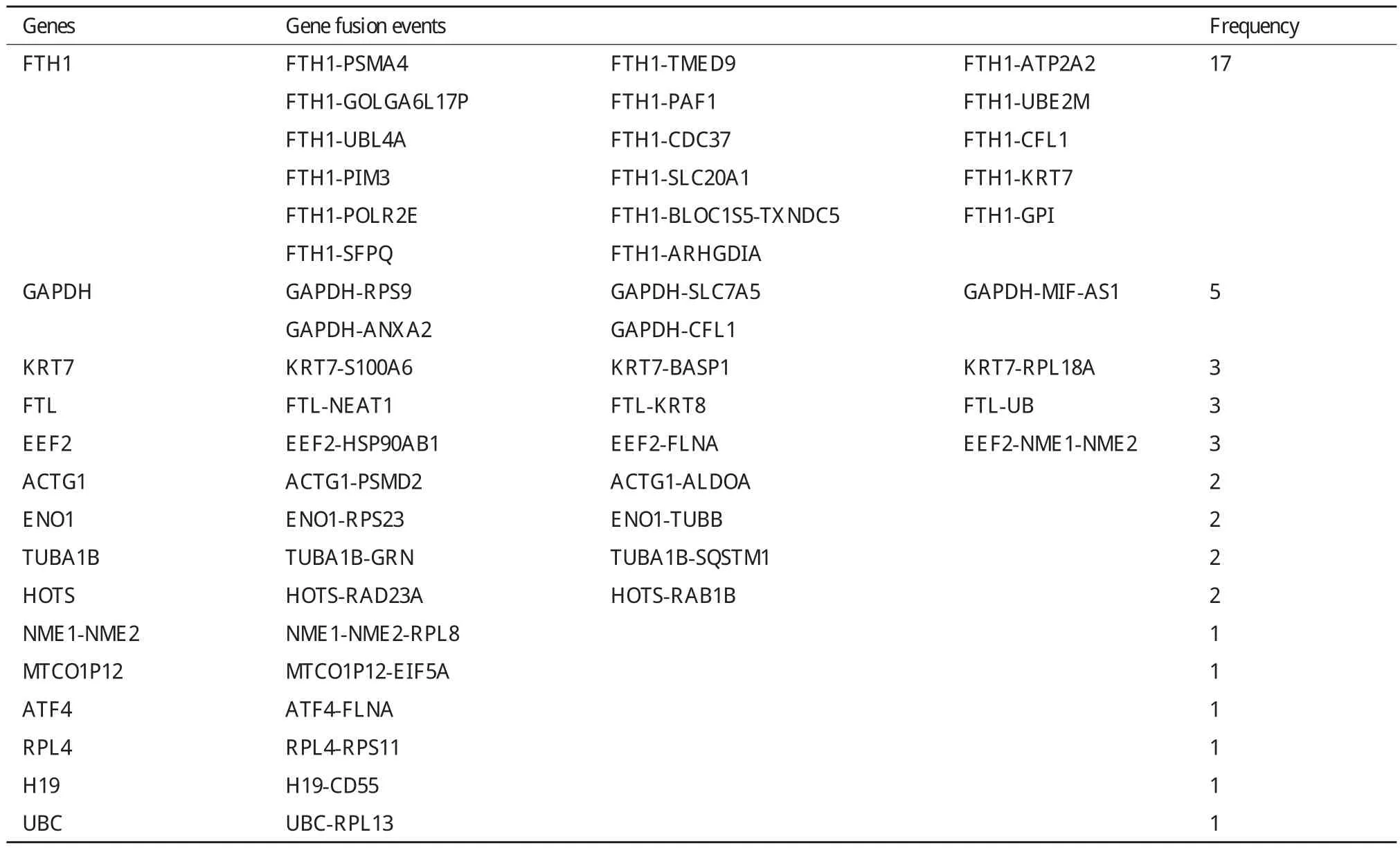
表1 基因融合事件統計
3 討論
本次轉錄組測序篩選得到顯著性差異表達基因有403個,其中上調的有269個而下調的有134個。GO和KEGG分析結果表明差異表達基因顯著參與了與腫瘤相關的生物學進程和信號通路。對5種主要的可變剪接類型進行統計,共得到可變剪接位點276個。本研究中一共得到SNP位點5 557個。在SNP位點中,非同義突變SNP位點有2 065個而同義突變SNP位點有3 492個,同義突變所占的比例在70%左右而非同義突變占30%左右,這與文獻報道的結果一致[13]。同時在SNP突變類型中,C/T和A/G突變的頻率最高,這可能是因為在人類基因組中存在大量的CpG島,同時CpG島中的C常常是甲基化的并容易發生脫氨基而轉變為T[14]。另外,我們這些獲取的SNP位點還存在一定的假陽性,如肝細胞癌HepG2細胞系經過長時間的培養,其染色體經常存在非整數倍的染色體變化并且包含如刪除和復制等的染色體改變,這些染色體的變化會直接影響SNP的檢測結果[15]。由于RNA-seq檢測的RNA在進行編輯的過程中也會導致序列變異從而可能被錯誤地注釋為SNP,因此一些檢測到的序列變異有可能是RNA編輯位點,而不是真正的SNP[16,17]。此外,由于無法保證對照組與處理組RNA樣本的完全一致,多肽9R-P201處理后導致的SNP中還會存在由于對照組與處理組RNA樣本不一致導致的SNP假陽性。對基因融合進行鑒定,共得到45個基因融合位點,其中FTH1基因與其它基因發生融合的次數有17次;研究發現FTH1基因在離子儲存和轉運起重要作用并調控細胞內離子內穩態[18]。
綜上所述,這些顯著差異表達的基因可能在多肽9R-P201激活的生物學功能和信號通路中起著重要的功能。同時可變剪接、SNP位點、基因融合等基因組結構的改變也暗示多肽9R-P201在這些事件上對肝癌HepG2細胞的調控影響。當然,9R-P201對于肝癌細胞的調控不僅局限于基因來發揮作用,通過發現更多的9R-P201下游調控因子以及信號通路將為基于9R-P201多肽為基礎的藥物開發將提供更多的支撐。
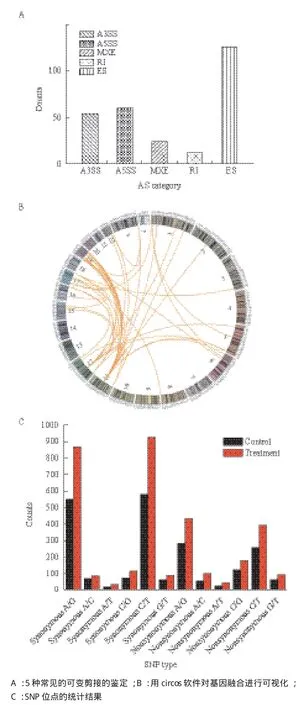
圖2 可變剪接、基因融合、SNP分析結果
4 結論
在肝癌發生發展過程中涉及多個與癌癥相關的生物學過程和信號通路的改變。本研究分析了肝癌HepG2細胞在9R-P201作用下基因差異表達情況、可變剪接、SNP位點和基因融合等。共得到可變剪接位點276個、SNP位點5 557個及基因融合位點45個。此外,還得到顯著差異表達的基因 403個,其中上調269個,下調134個。基因功能注釋結果表明差異表達基因富集上多個與腫瘤密切相關的生物學進程和信號通路,預示這些基因在9R-P201對肝癌HepG2細胞的調控過程中發揮重要的作用。
[1]Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide:sources, methods and major patterns in GLOBOCAN 2012[J]. Int J Cancer, 2015, 136(5):E359-386.
[2]Brito AF, Abrantes AM, Tralh?o JG, et al. Targeting hepatocellular carcinoma:what did we discover so far?[J]. Oncology Reviews, 2016, 10(2):302.
[3]Deng GL, Zeng S, Shen H. Chemotherapy and target therapy for hepatocellular carcinoma:New advances and challenges[J]. World Journal of Hepatology, 2015, 7(5):787-798.
[4]Meng Y, Gao X, Chen W, et al. Methionine enkephalin(MENK)mounts antitumor effect via regulating dendritic cells(DCs)[J]. International Immunopharmacology, 2017, 44:61-71.
[5]Xia L, Wu Y, Ma JI, et al. The antibacterial peptide from Bombyxmori cecropinXJ induced growth arrest and apoptosis in human hepatocellular carcinoma cells[J]. Oncology Letters, 2016, 12(1):57-62.
[6]Cui J, Huang J, Guo T, et al. Expression and selection of human foxm1c binding peptides and yheir inhibitions on MCF7 cancer cells[J]. Int J Pept Res Ther, 2014, 20(4):447-456.
[7]Bi Z, Liu W, D ing R, et al. A novel peptide, 9R-P201, strongly inhibits the viability, proliferation and migration of liver cancer HepG2 cells and induces apoptosis by down-regulation of FoxM1 expression[J]. Eur J Pharmacol, 2017, 796:175-189.
[8]Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq e xperiments with TopHat and Cufflinks[J]. Nature Protocols, 2012, 7(3):562-578.
[9]Kim D, Salzberg SL. TopHat-Fusion:an algorithm for discovery of novel fusion transcripts[J]. Genome Biol, 2011, 12(8):R72.
[10]Li HD, Menon R, Omenn GS, et al. The emerging era of genomic data integration for analyzing splice isoform function[J]. Trends in Gene tics:TIG, 2014, 30(8):340-347.
[11]Thomas PD, Mi H, Lewis S. Ontology annotation:mapping genomic regions to biological function[J]. Current Opinion in Chemical Biology, 2007, 11(1):4-11.
[12]Kanehi sa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment[J]. Nucleic Acids Res, 2008, 36:D480-484.
[13] Koberle B, Koch B, Fischer BM, et al. Single nucleotide polymorphisms i n DNA repair genes and putative cancer risk[J]. Archives of Toxicology, 2016, 90(10):2369-2388.
[14]Shastry BS. SNPs:impact on gene function and phenotype[J]. Methods in Molecular Biology, 2009, 578:3-22.
[15]Chen J, Driguneswar P, Shravan K, et al. Selecting c ell lines for SNP human identification assay development[J]. Atlas Journal of Biotechnology, 2011, 1(1):21-26, 2011.
[16]Gommans WM, Tatalias NE, Sie CP, et al. Screening of human SNP database identifies recoding sites of A-to-I RNA editing[J]. RNA, 2008, 14(10):2074-2085.
[17]Eisenberg E, Adamsky K, Cohen L, et al. Identification of RNA editing sites in the SNP database[J]. Nucleic Acids Research, 2005, 33(14):4612-4617.
[18]Guo J, Xu N, Yao Y, et al. Efficient expression of rec ombinant human heavy chain ferritin(FTH1)with modified peptides[J]. Protein Expression and Purification, 2016, 131:101-108.
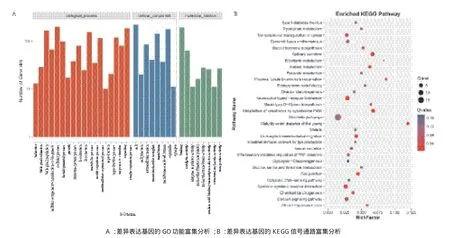
圖3 差異表達基因功能富集分析
(責任編輯 李楠)
Transcriptome Sequencing Analysis of Hepatocellular Carcinoma HepG2 Cells Induced by Antitumor Peptide 9R-P201
LIU Wen-rong1DING Ruo-fan1ZHANG Yi-ming1LI Yu-peng1LI Ling2GUO Zhi-yun1
(1. School of Life Science and Engineering,Southwest Jiaotong University,Chengdu 610031;2. Department of Pathology,the Third People’s Hospital of Chengdu,Chengdu 610031)
This wok aims to elucidate the regulation of 9R-P201 on hepatoma cells(HepG2)at transcriptome level by investigating the gene fusion,SNP(Single Nucleotide Polymorphism)mutation,alternative splicing and other events after 9R-P201 treating HepG2,and analyzing the biological processes and signaling pathways involved by differentially expressed genes. The expression differences of the genes before and after the 9R-P201 treating HepG2 cell line were detected by transcriptome sequencing. Meanwhile,gene fusion,SNPs,and alternative splicing were identified by tophat-fusion,SAMTOOLS software,and rMATS respectively. Functional enrichment analysis of differentially expressed genes were performed by GO(Gene Ontology)and KEGG(Kyoto Encyclopedia of Genes and Genomes). As results,276 alternative splicing events,5 557 SNP sites,and 45 gene fusion events were detected in the transcriptome sequencing. In addition,403 differentially expressed genes including 269 up-regulated and 134 down-regulated were detected. Gene GO and KEGG enrichment analysis showed that differentially expressed genes were significantly involved in the biological processes of cell growth,locomotion,as well as a number of cancer-related signaling pathways. In conclusion,the study revealed that the differentially expressed genes resulted from 9R-P201 treating HepG2 were significantly correlated with the biological processes and signaling pathways related to cancer and hepatocellular carcinoma HepG2 cell line,and a large number of alternative splicing events,SNP mutations,gene fusion events after 9R-P201 inducement occurred,suggesting this peptide may be used as a potential drug for subsequent hepatocellular carcinoma interventional therapy.
9R-P201;hepatocellular carcinoma;transcriptome sequencing;gene
10.13560/j.cnki.biotech.bull.1985.2017-0053
2017-01-26
中央高校基本科研業務費專項資金(2682016YXZT04),國家大學生創新性實驗計劃項目(201610613066)
劉文榮,男,碩士研究生,研究方向:非編碼RNA結構與功能;E-mail:liuzimu1992@gmail.com
郭志云,男,副教授,研究方向:生物信息學;E-mail:zhiyunguo@gmail.com
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
今日農業(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
現代出版(2020年3期)2020-06-20 07:10:34
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
天津醫科大學學報(2019年3期)2019-08-13 06:53:08
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
腫瘤預防與治療(2015年1期)2015-09-26 07:26:20
中國當代醫藥(2015年16期)2015-03-01 02:03:11
