親水作用色譜—質譜聯用法同時測定參附注射液中的14種有機酸
2017-05-17 01:21:49劉瑤張娜史社坡宋青青李軍宋月林
中國中藥雜志 2016年18期
劉瑤+張娜+史社坡+宋青青+李軍+宋月林+屠鵬飛
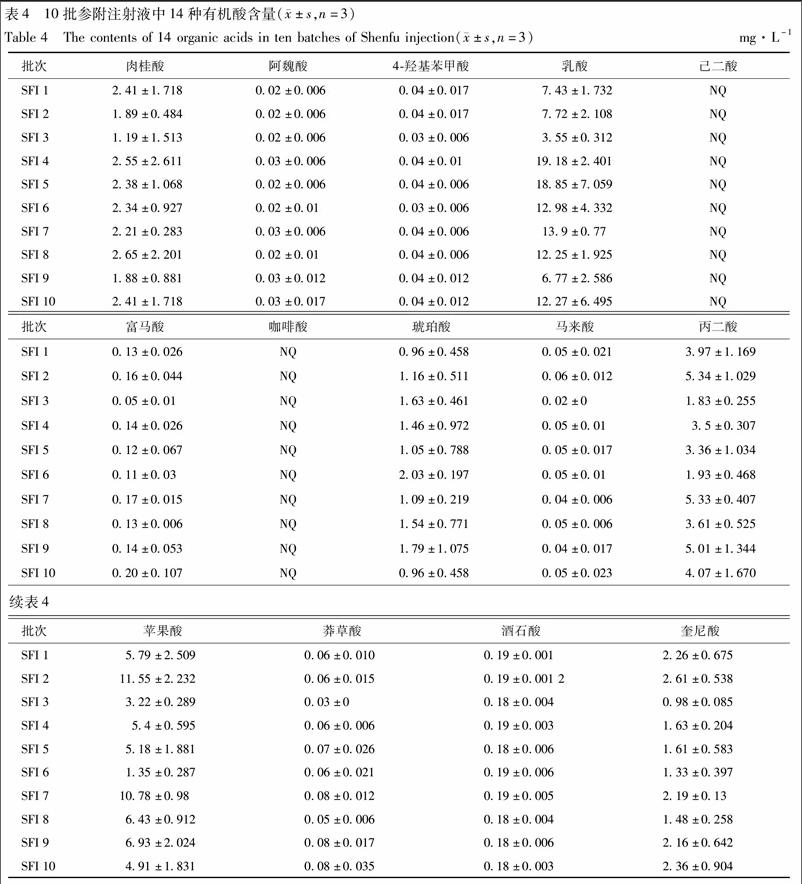

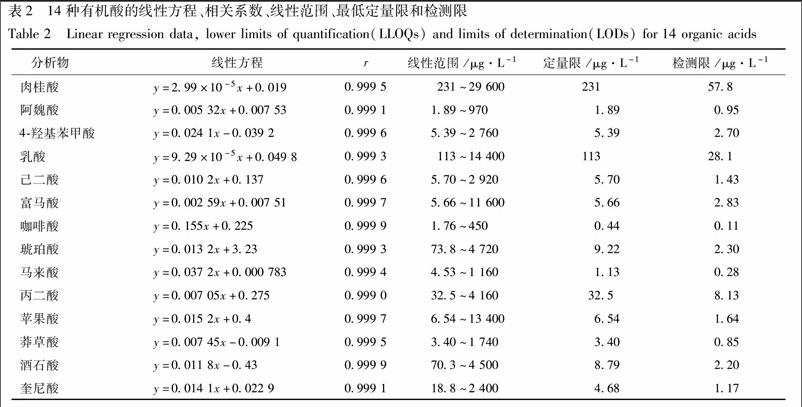
[摘要]有機酸廣泛分布在植物及相關產品中,其參與多種代謝途徑(如三羧酸循環),并表現出多種藥理活性。參附注射液作為廣泛使用的中藥注射劑,不良反應時有報道。因而,應深入闡明參附注射液的化學組成,并制定嚴格的質量標準,進而保障其臨床使用安全有效。參附注射液由紅參及黑順片經現代工藝提取制備而成,有機酸為其主要成分類別之一。該研究通過建立HTTJTC-LC-MS,同時測定參附注射液中肉桂酸、阿魏酸、4-羥基苯甲酸、乳酸、己二酸、延胡索酸、咖啡酸、琥珀酸、馬來酸、丙二酸、蘋果酸、莽草酸、酒石酸和金雞納酸等14種有機酸成分的含量。14種有機酸類成分在HTTJTC色譜柱上均獲得較好的保留和分離。除肉桂酸(231μg·L-1)、乳酸(113μg·L-1)和丙二酸(32.5μg·L-1)外,其余11個化合物的定量限均小于10μg·L-1。定量結果表明,蘋果酸、丙二酸、奎尼酸、乳酸和肉桂酸在參附注射液中含量較高(>1.89 mg·L-1),而10批樣品中均未檢測到咖啡酸和己二酸。該方法適用于參附等中藥注射液中多種有機酸含量的同時測定。
[關鍵詞]有機酸;參附注射液;多成分含量測定;親水作用色譜法;質譜
參附注射液由紅參和黑順片經現代工藝提取制備而成,具回陽救逆、益氣固脫之功效,為臨床常用中藥注射劑。主要用于陽氣暴脫的厥脫癥(感染性、失血性、失液性休克等)及陽虛(氣虛)所致的驚悸、怔忡、喘咳、胃疼、泄瀉、痹癥等作為廣泛使用的中藥注射劑。參附注射液的不良反應時有報道。因此,應深入闡明參附注射液的化學組成,并制定嚴格的質量標準。有機酸作為參附注射液的主要化學成分類別之一,在其藥效作用及不良反應中均發揮了重要作用。
有機酸廣泛分布于植物的根、葉、果實等部位。常見的植物有機酸,如脂肪族的酒石酸、草酸、蘋果酸、枸椽酸、抗壞血酸(維生素C)等和芳香族的水楊酸、咖啡酸等。有機酸常表現出抑菌、消炎、抗病毒、抗突變、抗癌、增強冠狀動脈血流量、降低食糜的pH、增加胰液分泌、改變腸道微環境等藥理活性。近年來,特別是在短鏈脂肪酸類化合物(SCFA)被認定為多種腸道及相關疾病的生物標志物(biomark-ers)后,有機酸的定性、定量分析受到了越來越多的關注。除少數芳香族有機酸外,有機酸類化合物僅在紫外檢測范圍的末端具有較弱的吸收。并且,該類化合物常表現出較強的極性,甚至是離子態,使得在常見的RP-C18色譜柱上很難實現較好的保留。這些因素給有機酸類化合物的準確定量帶來了極大的挑戰。目前,有機酸類化合物的分析主要采用離子色譜-質譜聯用(IC-MS)、液相色譜結合低pH流動相和液質聯用配備新型的五氟苯乙基(PFP)色譜柱等。然而,這些方法存在對設備要求高、靈敏度低、操作繁瑣等缺點,難以滿足復雜樣品中各有機酸類化合物快速、靈敏定量分析的需求。雖然親水作用色譜法(HILIC)已被廣泛報道適用于大極性化合物的色譜分析,HILIC用于有機酸的定量分析卻鮮有報道。因此,本研究擬充分利用HILIC的優點,建立快速、靈敏的HILIC-LC-MS方法,并將其用于復雜樣品的定量分析。
作為本課題組對參附注射液化學物質組系統研究的一部分,在對參附注射液中的氨基酸、核苷、烏頭生物堿、人參皂苷等成分進行了定性及定量分析的基礎上,本文進一步利用親水作用色譜法結合質譜(HILIC.LC-MS)同時測定參附注射液中14種有機酸類成分的含量。
1材料
島津SHIMADZU高效液相色譜系統,配備2個LC-20ADXR泵、SIL-20ACXR自動進樣器、CTO-20AC柱溫箱、SPD-M20A PDA檢測器、DGU-20A3在線脫氣機和CBM-20A控制器;ABSciex 5500 Qtrap質譜儀(美國ABSciex公司)配備ESI離子源和Analyst1.6.2軟件;Milli-Q超純水一體機(美國Millipore公司)。
14種有機酸對照品包括乳酸[L·(+)=lacticacid]、丙二酸(malonic acid)、馬來酸(maleic acid)、富馬酸(fumaric acid)、琥珀酸(succinic acid)、蘋果酸(D-malic acid)、己二酸(adipic acid)、莽草酸[(一).shikimic acid]、奎尼酸(quinic acid)、酒石酸(D-tartaric acid)均來自于SUPELC0的有機酸套裝(美國Sigma-Aldrich公司);咖啡酸(caffeic acid)和4.羥基苯甲酸(4-hydroxybenzoic acid)購自日本東京化成工業株式會社;阿魏酸(ferulic acid)購自上海阿拉丁生化科技股份公司;肉桂酸(cinnamic acid)購自北京國藥集團化學試劑有限公司;內標物為4.氟肉桂酸(IS),購自北京百靈威科技有限公司。乙腈(Optima LC-MS級別,美國Thermo-Fisher公司),甲酸(Optima LC/MS級別,美國Thermo-Fisher公司)。其余試劑均為分析純。
10批次的參附注射液(SFI1~SFI10)均由華潤三九藥業有限公司(四川雅安)提供。
2方法與結果
2.1LC-MS條件
色譜柱為Waters XBridge BEH Amide(4.6mmx150mm,3.5μm,美國Waters公司),柱溫40℃;0.1%甲酸.水溶液(A)和0.1%甲酸.乙腈(B)作為流動相;梯度洗脫(0~10.0min,98%-55%B;10.0~10.1min,55%~98%B;10.1-15.0min,98%B);流速0.8 mL·min-1;進樣體積2.0μL。
質譜檢測采用ESI負離子化模式;霧化氣N2(GSl)50 psi(1psi=6.89 kPa);輔助氣N2(GS2)50psi;氣簾氣(CUR)35 psi;碰撞氣(CAD)high;噴霧電壓(IS)-4500V;離子化溫度(TEM)550℃;選擇離子檢測模式(selected ion momtoring,SIM);射入電壓(EP)-10V;碰撞室射出電壓(CXP)-16V。各有機酸的定量離子對、保留時間及其他質譜參數(脫簇電壓DP和碰撞能CE)見表1。典型色譜圖見圖1。
2.2溶液配制
2.2.1對照品溶液制備取乳酸、丙二酸、琥珀酸、蘋果酸、4.羥基苯甲酸、肉桂酸適量,精密稱定,分別用DMSO配制成20mmol·L-1的對照品儲備液;取馬來酸、富馬酸、己二酸、莽草酸、奎尼酸、酒石酸、咖啡酸、阿魏酸適量,分別用DMSO配制成10mmol·L-1的對照品儲備液,4℃保存,備用。
取內標物4.氟肉桂酸(IS)適量,用DMSO配制成10 mmol·L-1的內標儲備液,再用40%乙腈逐步稀釋1000倍,配制成10μmol·L2的內標溶液,4℃保存,備用。
2.2.2供試品溶液制備吸取10批次參附注射液樣品各200μL,每批抽取3份,分別用40%乙腈稀釋5倍后,再吸取100μL再用10μmol·L-1的內標溶液稀釋10倍,用0.22μm微孔濾膜過濾,取續濾液,即得供試品溶液。
2.3線性關系考察
吸取適量各對照品的儲備液,混合,并用40%乙腈稀釋至1.0mL,配制成乳酸、丙二酸、馬來酸、富馬酸、琥珀酸、蘋果酸、4.羥基苯甲酸、己二酸、肉桂酸、莽草酸、咖啡酸、奎尼酸、阿魏酸、酒石酸質量濃度分別為144,41.6,11.6,116,94.4,268,55.2,29.2,296,34.8,36,192,38.8,45 mg·L-1的混標儲備液。用40%乙腈逐步倍半稀釋混標儲備液,獲得系列濃度梯度的混標溶液。分別吸取各濃度混標溶液100μL,用10μmol·L-1。的內標溶液稀釋10倍,即得系列濃度梯度的含內標的樣品溶液。在優化的實驗條件下,對含有相同濃度內標的一系列標準溶液按照2.1項下條件進行檢測,重復進樣3次。以各有機酸峰面積與內標峰面積的比值作為縱坐標(Y),各有機酸質量濃度(μg·L-1)作為橫坐標(X),繪制標準曲線,進行線性回歸得標準曲線。對標準溶液進行逐步稀釋,分別取信噪比(S/N)約為3和10時各分析物的濃度作為檢測限(μg·L-1,LOD)和最低定量限(μg·L-1,LLOQ)。14種待測成分的線性回歸方程、線性范圍、相關系數(r)、檢測限和最低定量限見表2。
2.4精密度試驗
分別選擇高、中、低濃度標曲樣品溶液作為質控樣品,具體濃度見表3。取3個濃度的質控樣品進行日內(intra-day)和日間(inter-day)精密度考察。按2.1項下LC-MS條件進行樣品分析,日內精密度通過每隔2h進樣1次,共進樣6次考察;日間精密度通過連續3d,1 d3次,采用同法測定考察,計算各有機酸含量的相對標準偏差(RSD)。由表3可見各化合物的日內精密度RSD≤13%,日間精密度RSD≤15%,均符合方法學的要求。
2.5重復性試驗
取參附注射液1批次,按2.2.2項下方法平行制備6份,測定峰面積,并計算各待測化合物的含量,RSD為2.1%~8.6%,結果表明該方法的重復性良好。
2.6穩定性試驗
取同一份供試品溶液,室溫放置,分別于0,2,4,8,12,18,24 h,按2.1項下LC-MS條件進行樣品分析,計算各待測化合物含量的相對標準偏差,各RSD為3.7%~10%,表明各待測化合物在室溫下24 h之內能夠穩定存在。
2.7加樣回收率試驗
取已測得含量的參附注射液樣品,分別精密加入適量體積的高、中、低3個濃度的混標溶液,見表3,按2.2.2項下方法處理后按2.1項下LC-MS條件測定,每一濃度平行測定3次,計算平均回收率。由表3可見,除2種未檢測到的有機酸(己二酸和咖啡酸)外,其余12種有機酸的平均加樣回收率為86.33%一114.3%(RSD≤14%),表明加入不同濃度的混標溶液后,各分析物的回收率較為穩定,準確度較好。
2.8樣品溶液含量測定
將10批參附注射液按2.2.2項下供試品溶液制備方法制備,依照2.1項下色譜和質譜條件測定,重復3次。將所得峰面積代入標準曲線,計算各批次參附注射液中各有機酸類成分的含量。典型圖譜見圖1B,測定結果見表4。
3討論與結論
有機酸廣泛分布于中藥材及中藥復方制劑當中。由于苯環等強發色團的缺失,除少數芳香族有機酸外,該類化合物只具備較弱的紫外吸收。另一方面,由于羧基的存在,有機酸類化合物常表現出較強的極性。因此,常用的HPLC-UV配備RP-C18色譜柱的方法難以實現有機酸的全面準確定量分析。本文采用HILIC色譜柱實現了有機酸類分析物的較好保留,并且采用質譜檢測器,利用選擇離子監測(SIM)模式對各有機酸類化合物進行靈敏地檢測,建立了參附注射液中14個有機酸類分析物同時定量分析的方法。實驗過程中篩選了多種HILIC色譜柱,方法學考察和實際樣品測定結果表明,使用化學性質穩定的三鍵鍵合的酰胺基鍵合相的Waters XBridge BEH Amide色譜柱對有機酸類化學成分保留能力強、柱流失低,能夠實現多種有機酸的快速分離。已有文獻報道常通過調低流動相pH(硫酸或磷酸鹽緩沖體系,pH<3)流動相,對色譜柱損害較大。并且,由于非揮發性緩沖鹽的使用,使得無法采用蒸發光散色檢測器及質譜檢測器。而本文采用0.1%甲酸作為流動相改性劑,不僅能很好地降低色譜柱的損害,而且能滿足質譜檢測器的要求。在對待測化合物離子對進行篩選的過程中,發現有機酸類分析物由于結構中存在羧基,響應最好的離子對一般為[M-H]->[M-H-CO2]-,能夠很好地與其他類型化合物進行區分。
附子生物堿及人參皂苷類成分通常被認為是參附注射液中的主要活性成分。然而,在系統研究過程中發現參附注射液中大量存在各種大極性成分,主要包括有機酸類、氨基酸類、核苷類等化學類型。本研究在定量分析參附注射液中附子生物堿、人參皂苷、氨基酸及核苷類成分的基礎上,進一步建立了一種可快速測定參附注射液中14種有機酸含量的色譜.質譜聯用法。在所測的10批參附注射液中,蘋果酸、丙二酸、奎尼酸、乳酸和肉桂酸的含量均較大(>1.89mg·L-1),而咖啡酸和己二酸均未檢測到。本文的定量分析結果對于參附注射液的系統質量控制及用藥安全標準提升具有重要意義。同時,所建立的HILIC-LC-MS方法適用于參附等中藥注射劑中多種有機酸含量的同時測定。
