漁藥藥效學專題講座
——第一章 漁藥藥物效應動力學基礎(3)
2017-03-20 03:44:00汪建國
漁業致富指南 2017年3期
關鍵詞:效應
汪建國
(中國科學院水生生物研究所 研究員 博導)
漁藥藥效學專題講座
——第一章 漁藥藥物效應動力學基礎(3)
汪建國
(中國科學院水生生物研究所 研究員 博導)
(二)藥物-受體學說
1.占領學說 C1ark于1926年提出的占領學說認為:藥物效果與藥物占領受體數目成正比,藥物占領受體數目取決于細胞表面受體的密度和受體周圍的藥物濃度,當全部受體被占領,即達到Emax。占領學說只適用于激動藥,但不能解釋2個激動藥激動同樣受體而產生不同的Emax。
1954年Ariens修正了占領學說,他把決定藥物與受體結合時產生效應的能力稱為內在活性。藥物與受體結合不僅需要親和力,而且還需要內在活性才能激動受體而產生效應。只有親和力而沒有內在活性的藥物,雖可與受體結合,但不能激動受體故不產生效應。
1956年Stephenson又提出,藥物只占領小部分受體即可產生最大效應,未經占領的受體稱為儲備受體。因此,當不可逆性結合或其它原因而喪失一部分受體時,并不會立即影響最大效應。進一步研究發現,內在活性不同的同類藥物產生同等強度效應時,所占領受體的數目并不相等。激動藥占領的受體必須達到一定閾值后才開始出現效應。當達到閾值后被占領的受體數目增多時,激動效應隨之增強。閾值以下被占領的受體稱為沉默受體。
2.速率學說 1961年Paton 提出了受體速率學說。該學說認為影響藥物效應的大小的主要因素不是受體被占領數量的多少,而是藥物分子與受體的結合速率。認為每當一個藥物分子和受體相碰撞時即可產生一定量的刺激,并能被傳到相應效應器而產生效應。使用這一學說的簡單之處在于,僅以受體-藥物結合和解離速率參數即可推算結果,無須使用內在活性和效能等參數。但問題在于這一學說無法解釋藥物與受體多種類型的相互作用。故這一學說被使用的范圍并不大。
3.二態模型學說 Kar1in A.和Changenx JP.分別提出了藥物受體作用的兩態學說,又稱變構學說。它們都認為受體存在活性狀態(R*)和非活性狀態(R)。兩者均可與藥物結合,而且活性和非活性受體之間也可以相互轉化,二者處于動態平衡。激動劑主要與R*結合形成R*A,而拮抗劑則主要形成RB。實際上激動劑和拮抗劑分別推動R*和R的轉換向著各自方向移動。這兩種狀態的受體之比例及兩種狀態受體與藥物的親和力大小決定著生物效應的大小。這種模型考慮到了受體與藥物間相互作用導致的受體活性改變,更接近于實際的藥物-受體反應狀況,但用以直接說明藥物與效應之間的定量關系尚不完備。
4.誘導契合學說 Kosh1and等提出的誘導契合學說,可以較好地解釋配基與受體結合的實際過程。即藥物與受體蛋白結合時,可誘使受體的空間構象發生可逆改變,而且這種結構變化即可導致生物效應。而且這種學說也可以解釋藥物與受體之間的協作效應。
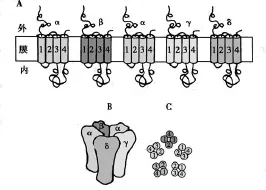
圖7 N2型乙酰膽堿受體陽離子通道分子結構示意圖
需要注意的是,上述這些受體學說大多只能在某種特定的條件或范圍內解釋受體與藥物的作用和受體與效應之間的關系,均存在一定的局限性。
(三)受體的類型、分布與特性
藥物與受體結合形成復合物,引起一系列細胞反應,從而引起最終效應。不同的受體所處位置、本身結構以及信息傳導過程各有不同,所引發的最終效應也都不一樣。
1.離子通道受體 存在于快速反應細胞膜上的受體,多為含離子通道的受體。離子通道受體的主要特征是:受體蛋白本身組成一個跨膜的離子通道(圖7),通道的開或關控制一些離子的跨膜流量,并通過改變細胞內離子濃度影響細胞功能。通道的開關則由配基與受體的結合或解離控制。例如神經肌肉接頭處的N2膽堿受體是由5個亞基在細胞膜內呈五邊形排列圍成的鈉離子通道,當乙酰膽堿與受體結合后,鈉通道開放,鈉離子內流增加,膜去極化,肌肉開始收縮。
2.G蛋白偶聯受體,間接影響離子通道或第二信使 這是目前已發現的種類最多的受體類型,其激動劑種類包括生物胺、蛋白激素、多肽激素、腦/腸多肽、花生四烯酸系列的活性物質,淋巴細胞活性因子等。這類受體位于細胞膜,都由一條肽鏈形成,其N-末端在細胞膜外,為配體的結合部位;C末端在細胞內,為G蛋白結合部位。而且肽鏈形成7個跨膜螺旋結構和相應的三個細胞外和三個細胞內環,所以這類受體又稱為7跨膜區受體(7TM)。7TM受體不僅具有共同的結構特征,而且具有共同的細胞內信號放大方式。這種結構能夠穩定G蛋白結構,使它們能被脂質雙分子層緊緊固定并保證它們不能進入到胞質中去(圖8)。
G蛋白是鳥苷酸結合調節蛋白的簡稱,存在于細胞膜內側,由α、β、γ三個亞單位組成的三聚體,靜息狀態時與GDP結合。當受體與配體結合后,相應受體被激活,GDP-α、β、γ復合物在Mg2+參與下,結合的GDP與胞漿中GTP發生交換,GTP-α與β、γ分離并與相應的效應機制結合,同時配體與受體分離。α亞單位內在的GTP酶促使GTP水解為GDP,激活效應機制,從而恢復原來的靜息狀態。G蛋白主要有兩類,一類為興奮性G-蛋白(GS),激活腺苷酸環化酶(AC)使cAMP增加;另一類為抑制性G-蛋白(Gi),抑制AC,使cAMP減少。G-蛋白還介導心鈉素及NO對鳥苷酸環化酶(GC)的激活作用。此外,G-蛋白對磷脂酶C、磷脂酶A2,Ca2+、K+離子通道等有重要調節作用。一個受體可激活多個G-蛋白,一個G-蛋白可以轉導多個信號給效應器(effector),從而調節神經、心肌、平滑肌等多種細胞的功能。

圖8 G蛋白偶聯受體的結構
3.跨膜激酶活性受體,直接調節蛋白磷酸化 這一家族包括許多多肽激素和生長因子的受體,例如上皮生長因子、血小板生長因子及某些淋巴因子等。它們位于細胞膜,只有一個疏水的跨膜區段,包括細胞膜外結合域、跨膜區、細胞內酶活性區幾個部分。受體分子本身不具有激酶活性,當激動劑與細胞膜外的識別部位結合后,其細胞內某些可溶性酪氨酸激酶被激活,首先在特定的部位發生自身磷酸化,然后再將磷酸根轉移到其效應器上,使效應器蛋白的酪氨酸殘基被磷酸化,從而改變效應器的活性(圖9)。
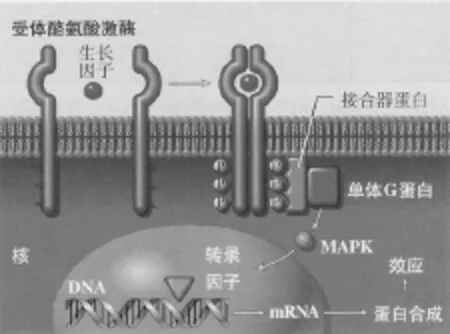
圖9 跨膜激酶活性受體作用機制
4.核受體,對DNA的轉錄進行調節大多數甾體激素類受體屬于這一類型,如性腺激素受體(雄激素受體、雌激素受體、孕激素受體)、腎上腺皮質激素受體(糖皮質激素受體、鹽皮質激素受體)、甲狀腺激素受體、維甲酸受體、維生素D3受體等。這類受體位于細胞內,主要是在細胞核上,細胞漿中也能檢測到該類受體。這類受體和核內特定的DNA結合,并通過這種結合影響遺傳信息的轉錄。當其被激活后,可通過對DNA轉錄的調節影響蛋白質的合成而導致生理或生化反應。例如腎上腺皮質激素激動受體所產生的抗炎作用就是這種機制。作用于細胞內受體的內源性配基或外源性配基都必須透過細胞膜后才能起作用,因此要求配基具有一定的脂溶性(圖10)。
(四)受體與藥物的結合
1.親和力與內在活性 藥物與受體結合形成復合物而引起效應,效應強弱遵循質量作用定律,即藥物作用強弱與藥物占領受體數目多少成正比,可用下列公式表示:
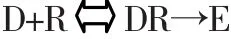
式中D代表藥物,R代表受體,DR代表藥物受體復合物,E代表效應。藥物與受體結合后產生生理效應,不僅需要有親和力,而且還需要有內在活性。親和力是指藥物與受體結合的能力,它反映出藥物的作用強度(效價強度);內在活性是藥物與受體結合后激動受體產生效應的能力,它反映出藥物的最大效應(效能)。當親和力相等,藥物最大效應取決于內在活性;當內在活性相等時,藥物作用強度取決于親和力的大小。親和力一般受采用藥物-受體復合物的解離常數(KD)的倒數(1/KD)來表示,KD是引起最大效應一半時(即50%受體被占領)的藥物劑量。
2.作用于受體的藥物分類 根據藥物與受體間親和力與內在活性的不同,將藥物總體分為激動藥和拮抗藥兩大類
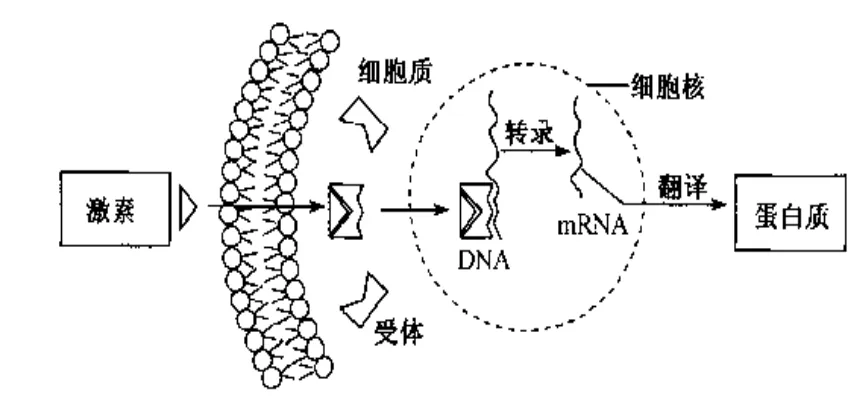
圖10 核受體作用機制
(1)激動藥 對受體既有親和力又有內在活性的藥物,能與受體結合并產生效應。根據親和力和內在活性不同,激動藥可分為:
①完全激動藥,是具有較強親和力和內在活性(α=1)的藥物;
②部分激動藥,是對受體有較強的親和力,但內在活性較弱(0<α<1)的藥物,單獨應用,有較弱的效應,可與激動藥競爭受體;
③負性激動藥,也稱反轉激動藥,激動受體后產生激動藥相反的效應。負性激動藥與受體結合后使受體構型發生質的改變,引起與激動藥相反的效應。
(2)拮抗藥 也稱阻斷藥,是對受體有較強親和力,而無內在活性(α=1)的藥物。根據藥物與受體的結合的可逆性,拮抗藥可分為:
①競爭性拮抗藥,由于與受體的結合是可逆的,可與激動藥競爭與受體結合;
②非競爭性拮抗藥,由于與受體的結合是牢固和相對不可逆的,或結合后引起受體構型改變和活性降低,使激動藥無法完全對抗這種非競爭性改變,因而效應降低。
3.受體與藥物的相互作用
(1)競爭性拮抗 競爭性拮抗發生在:①閾劑量增加;激動藥與拮抗藥之間;②激動藥與部分激動藥之間;③拮抗藥或部分激動藥與內源性激動劑(遞質、激素、內在活性物質)之間。由于激動藥、拮抗藥和部分激動藥對受體都具有親和力,可相互競爭同一受體,發生競爭性結合或拮抗。它們與受體的結合是可逆的,合并用藥的效應取決于各自藥物的濃度和親和力。觀察拮抗藥或部分激動藥對激動藥量-效曲線的影響是研究競爭性拮抗的常用方法。通常,先做激動藥的量-效曲線,把激動藥從受體部位清洗干凈后,加入固定濃度的拮抗藥或部分激動藥,再觀察在拮抗藥或部分激動藥存在下激動藥的量效曲線的變化(圖11)。
激動藥與拮抗藥的競爭性拮抗激動藥有強的內在活性,與受體結合可引起藥理效應;拮抗藥無內在活性,與受體結合后不產生效應。拮抗藥存在時,激動藥量效曲線發生下列變化:
②曲線平行右移(斜率不變),KD值增加;

圖11 不同藥量的競爭性拮抗劑、非競爭性拮抗劑、激動劑和部分激動劑相互作用量效關系圖
③Emax不變。根據拮抗藥對激動藥量效曲線的影響,可測定拮抗藥的pA2值,或是使激動藥的KD值增加一倍時拮抗藥摩爾濃度的負對數。pA2是使激動藥濃度提高一倍時所產生的效應與原來的效應相同時所用拮抗藥摩爾濃度的負對數,是衡量拮抗藥親和力的指標。pA2越大,拮抗藥親和力越強,對激動藥的阻斷作用越明顯。
激動藥與部分激動藥的競爭性拮抗部分激動藥存在時使激動藥的量效曲線發生下列變化:
①閾劑量減小;
②在激動藥劑量較小時,量效曲線左移,說明部分激動藥起激動藥的作用;
③在激動藥劑量較大時,量效曲線右移,說明部分激動藥起拮抗藥的作用;④Emax不變。
(2)非競爭性拮抗 有些拮抗藥與受體結合牢固,形成的復合物解離速度很慢,以致造成不可逆性結合,此時,增大激動藥的濃度也無法達到原來的Emax。
非競爭性拮抗可使激動藥的量效曲線發生下列變化:
①曲線非平行右移;
②Emax降低.根據非競爭性拮抗藥使最大效應降低的程度,可測得其pD2′值。pD2′是指使激動藥最大效應降低一半所需非競爭性拮抗藥的摩爾濃度的負對數,是衡量非競爭性拮抗藥親和力的參數。
(五)受體的調節
受體雖是遺傳獲得的固有蛋白,但并不是固定不變的,而是經常處于代謝轉換的動態平衡中,受體的數目、與配體的親和力以及效應經常受內環境變化以及藥物的影響而發生改變。各種因素使細胞上受體發生質和量變化的過程稱為受體調節。受體調節的生理意義在于通過調節使機體能更好地適應內外環境的變化,例如受體反應性減弱可保護細胞免受過量或長期刺激而導致生理功能紊亂,但受體調節過度則又會引起一系列病理性的后果。
1.受體失敏 在與配基作用一段時間后,受體對配基的敏感性和反應性下降的現象稱為失敏。失敏可以由于受體數目的減少和/或由于受體與配基親和力降低所致,它通常具有劑量依賴性、時間依賴性和可逆性等特點。
失敏又可分為以下兩種情況:失敏如果發生在與配基特異性結合的受體上,稱為同種失敏,即某種受體被配基激活后引起的失敏僅為該受體本身,而同一細胞上的其他受體系統并無實質性變化。
失敏如果發生于配基的非特異的受體上,即某種激動劑在作用一段時間后,不僅使其特異性受體對其反應降低,還使同一細胞上其他受體對它們各自激動劑的反應減弱。例如長期在與去甲腎上腺素接觸后,細胞不 僅對去甲腎上腺素的反應性降低(同種失敏),而且對前列腺素的反應性也降低(異種失敏)。
2.受體增敏 增敏是與失敏相反的一種現象,即當配基與受體作用一段時間后,受體的數目增加和/或親和力增加的現象。增敏也可發生于同種受體或異種受體,例如長期使用β腎上腺素受體阻斷劑的情況下突然停藥可出現受體敏感性比正常增高(同種增敏);又如大鼠長期飼以甲狀腺素后可出現心肌中β腎上腺素受體的合成增加,結合位點增加(異種增敏)。
(未完待續)
(通聯:430072,中國科學院水生生物研究所 武漢市武昌東湖南路7號)
猜你喜歡
核科學與工程(2021年4期)2022-01-12 06:30:26
今日農業(2020年19期)2020-12-14 14:16:52
小學生必讀(中年級版)(2020年9期)2020-12-04 02:07:22
科學大眾(2020年17期)2020-10-27 02:49:10
紅土地(2018年11期)2018-12-19 05:10:56
意林·全彩Color(2018年9期)2018-11-13 22:49:38
中學物理·高中(2016年12期)2017-04-22 11:53:03
中國衛生(2016年4期)2016-11-12 13:24:14
中國衛生(2014年4期)2014-12-06 05:57:14
小櫻桃·童年閱讀(2014年11期)2014-12-01 22:21:30
