阿膠三寶膏的質量標準提高研究Δ
2017-03-03 06:45:13林永強徐麗華山東省食品藥品檢驗研究院濟南250101
中國藥房 2017年3期
焦 陽,汪 冰,林永強,徐麗華(山東省食品藥品檢驗研究院,濟南 250101)
阿膠三寶膏的質量標準提高研究Δ
焦 陽*,汪 冰,林永強,徐麗華(山東省食品藥品檢驗研究院,濟南 250101)
目的:提高阿膠三寶膏的質量標準。方法:采用超高效液相色譜-質譜聯用法(UPLC-MS)鑒別制劑中的阿膠:色譜柱為Acquity UPLC BEH-C18,流動相為乙腈-0.1%甲酸溶液(梯度洗脫),流速為0.3 mL/min,進樣量為5μL,離子源為電噴霧離子源,毛細管電壓為3.5 kV,毛細管出口電壓為120 V,錐孔電壓為50 V,脫溶劑溫度為400℃,干燥氣流速為10 L/min,霧化器壓為40 psi,碰撞能量為10~45 V,掃描范圍m/z 200~1 500,檢測方式為正離子模式(ESI+),多反應監測(MRM);采用高效液相色譜法測定制劑中4種水解氨基酸的含量:色譜柱為Agela Venusil XBP-C18,流動相A為乙腈-0.1 mol/L乙酸鈉溶液(36%乙酸調pH至6.5)(7∶93,V/V),流動相B為80%乙腈溶液(梯度洗脫),流速為1.0 mL/min,檢測波長為254 nm,柱溫為43℃。結果:阿膠的UPLC-MS圖峰形清晰,專屬性強,陰性無干擾。L-羥脯氨酸、甘氨酸、丙氨酸、L-脯氨酸檢測進樣量線性范圍分別為0.081 9~0.655 2 μg(r=0.999 9)、0.186 2~1.489 6 μg(r=0.999 8)、0.070 3~0.562 0 μg(r=0.999 9)、0.124 2~0.993 6 μg(r=0.999 9);精密度、穩定性、重復性試驗的RSD<2.0%;加樣回收率分別為99.85%~103.69%(RSD=1.35%,n=6)、99.91%~103.93%(RSD=1.46%,n=6)、96.86%~101.27%(RSD=1.69%,n=6)、97.44%~101.45%(RSD=1.54%,n=6)。結論:提高的標準能更加有效地控制阿膠三寶膏的質量。
阿膠三寶膏由阿膠、大棗和黃芪3味中藥組合而成,具有補氣血、健脾胃之功效,常用于氣血兩虧、脾胃虛弱所致的心悸、氣短、崩漏、浮腫、食少等癥的治療[1]。其最早載于《衛生部藥品標準·中藥成方制劑(第二冊)》[2],但僅收載了性狀和常規檢查項。現行標準為2015年版《中國藥典》(一部),增加了采用高效液相色譜法(HPLC)測定黃芪甲苷含量和總氮量項目。本試驗對現行標準進行完善,增加了制劑中君藥阿膠的特征鑒別[3],并將原標準中總氮量的測定修訂為阿膠中4種水解氨基酸的HPLC定量測定[4]。
1 材料
1.1 儀器
Quattro Premier XE型超高效液相色譜-質譜聯用(UPLC-MS)儀、LC-20A型HPLC儀(包括LC-20AT四元泵、SPD-20A紫外可見光檢測器、DGU-20A5自動脫氣裝置、SIL-20AC自動進樣器、CTO-20AC柱溫箱)均購自美國Waters公司;CP225D型電子分析天平(德國Sartorius公司);KQ5200DE型數控超聲波清洗器(昆山市超聲儀器有限公司,功率:500 W,頻率:40 kHz)。
1.2 藥品與試劑
阿膠三寶膏(山東東阿阿膠股份有限公司,批號:120701、1211001、1301001,規格:250 g/瓶);L-羥脯氨酸對照品(批號:111578-200201,純度:100%)、甘氨酸對照品(批號:140689-201103,純度:99.9%)、丙氨酸對照品(批號:140680-201303,純度:99.9%)、L-脯氨酸對照品(批號:140677-201206,純度:100%)、阿膠對照藥材(批號:121274-201202)均購于中國食品藥品檢定研究院;胰蛋白酶(美國Promega公司,批號:ADV5280000 01120296);乙腈、甲醇為色譜純,其余試劑均為分析純,水為純化水。
2 方法與結果
2.1 阿膠的鑒別
2.1.1 試驗條件 (1)色譜條件。色譜柱:Acquity UPLC BEH-C18(100 mm×2.1 mm,1.7 μm);流動相:乙腈(A)-0.1%甲酸溶液(B),梯度洗脫(0~25 min,5%→20%A;25~40 min,20%→50%A);流速:0.3 mL/min;進樣量:5μL。(2)質譜條件。離子源:電噴霧離子源;毛細管電壓:3.5 kV;毛細管出口電壓:120 V;錐孔電壓:50 V;脫溶劑溫度:400℃;干燥氣流速:10 L/min;霧化器壓:40 psi;碰撞能量:10~45 V;掃描范圍:m/z 200~1 500;檢測方式:正離子模式(ESI+),多反應監測(MRM)。
2.1.2 對照藥材溶液的制備 取阿膠對照藥材0.1 g,加1%碳酸氫銨溶液50 mL,超聲處理30 min,微孔濾膜濾過,取續濾液100μL,置于微量進樣瓶中,加胰蛋白酶溶液(胰蛋白酶100μg溶于1%碳酸氫銨溶液100μL中,下同)10μL,搖勻,37℃恒溫酶解12 h,即得。
2.1.3 供試品溶液的制備 取樣品1.0 g,加1%碳酸氫銨溶液50 mL,超聲處理30 min,微孔濾膜濾過,取續濾液100μL,置于微量進樣瓶中,加胰蛋白酶溶液10μL,搖勻,37℃恒溫酶解12 h,即得。
2.1.4 陰性對照溶液的制備 按阿膠三寶膏處方和制備工藝制備缺阿膠的陰性樣品,并按“2.1.3”項下方法制成陰性對照溶液。
2.1.5 專屬性試驗 取“2.1.2”“2.1.3”“2.1.4”項下對照藥材溶液、供試品溶液、陰性對照溶液各適量,按“2.1.1”項下試驗條件進樣測定,記錄色譜,詳見圖1。結果表明,陰性對照在相應位置未見干擾峰,方法專屬性良好。
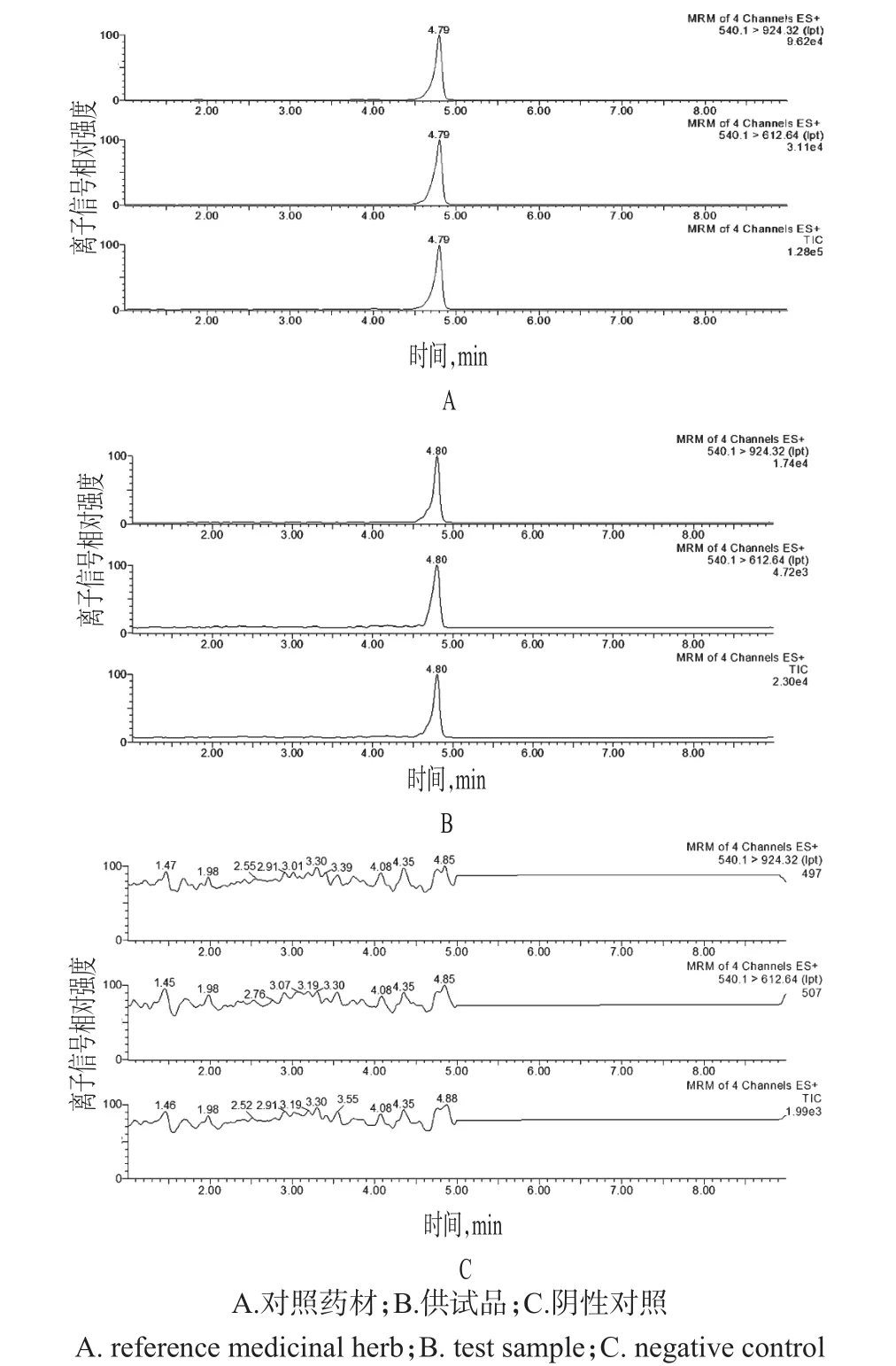
圖1 對照藥材、供試品、陰性對照的超高效液相色譜-質譜圖Fig 1 UPLC-MS chromatograms of reference medicinal herbs,test samples and negative control
2.1.6 精密度試驗 取“2.1.2”項下對照藥材溶液適量,按“2.1.1”項下試驗條件連續進樣測定6次,記錄峰面積。結果,m/z 539.8→612.4提取離子流峰與m/z 539.8→923.8提取離子流峰峰面積的RSD分別為4.32%、4.18%,表明儀器精密度良好。
2.1.7 樣品鑒別 取3批樣品各適量,分別按“2.1.3”項下方法制備供試品溶液,再按“2.1.1”項下試驗條件進樣測定,鑒別其成分,詳見圖2。結果,3批樣品的UPLCMS出峰時間一致。
2.2 4種水解氨基酸的含量測定
2.2.1 色譜條件 色譜柱:Agela Venusil XBP-C18(250 mm×4.6 mm,5 μm);流動相A:乙腈-0.1 mol/L乙酸鈉溶液(36%乙酸調pH至6.5)(7∶93,V/V),流動相B:80%乙腈溶液,梯度洗脫(0~20 min,100%→93%A;20~23.9 min,93%→88%A;23.9~24 min,88%→85%A; 24~39 min,85%→66%A;39~40 min,66%→0A);流速:1.0 mL/min;檢測波長:254 nm;柱溫:43℃。
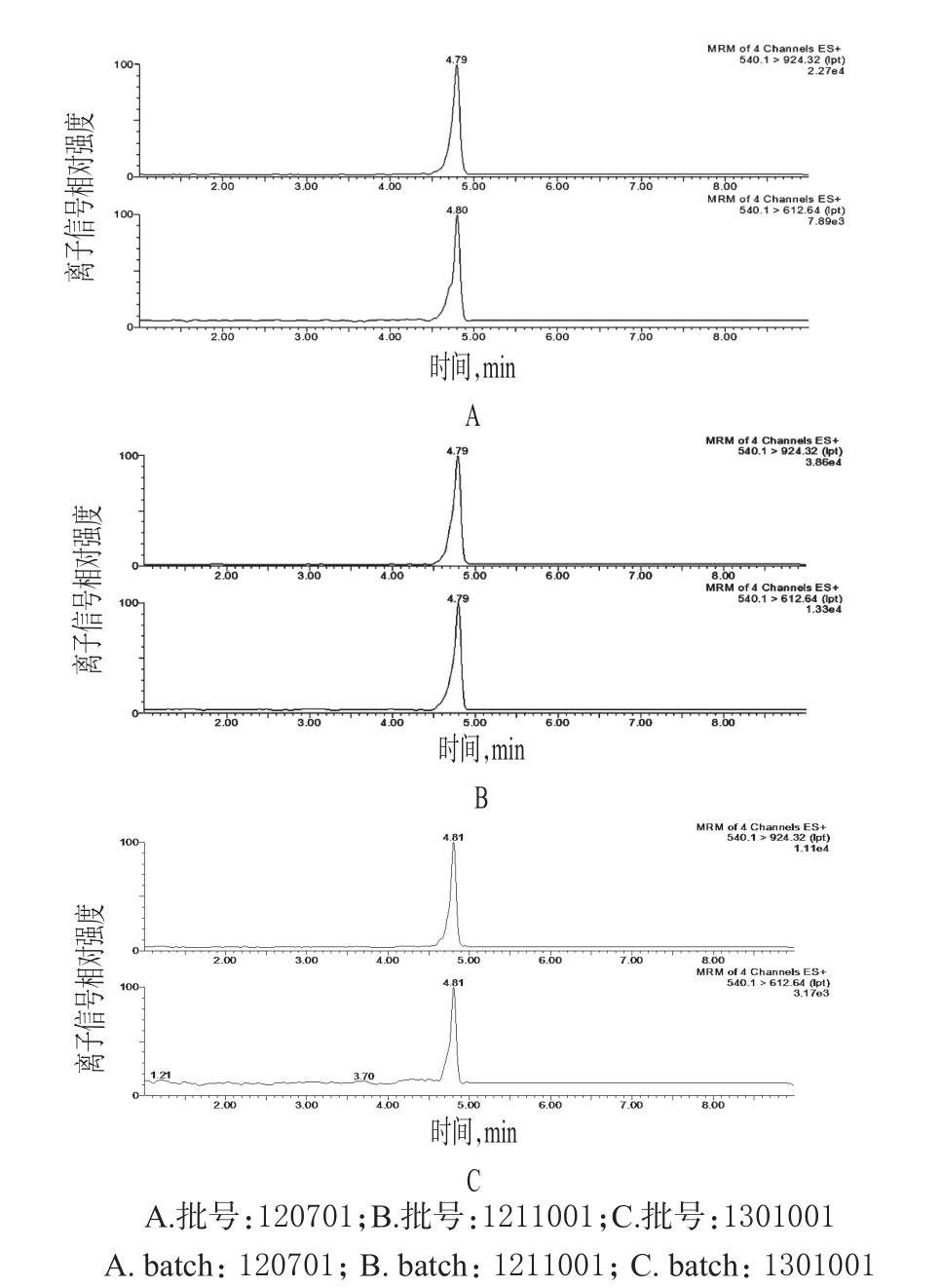
圖2 樣品的超高效液相色譜-質譜圖Fig 2 UPLC-MS chromatograms of samples
2.2.2 混合對照品溶液的制備 取待測水解氨基酸對照品適量,加0.1 mol/L鹽酸溶液制成L-羥脯氨酸、甘氨酸、丙氨酸、L-脯氨酸質量濃度分別為0.081 9、0.186 2、0.070 3、0.124 2 mg/mL的混合對照品溶液。
2.2.3 供試品溶液的制備 取樣品約2.5 g,精密稱定,加0.1 mol/L鹽酸溶液20 mL,超聲處理30 min,放冷,加0.1 mol/L鹽酸溶液定容至25 mL,搖勻。精密量取上述溶液2 mL,置于10 mL安瓿中,加2 mL鹽酸,熔封,置于150℃恒溫箱中1 h,放冷,移至蒸發皿中,用水10 mL分次洗滌,洗液并入蒸發皿中,蒸干,殘渣加0.1 mol/L鹽酸溶液溶解并定容至25 mL,搖勻,濾過,即得。
2.2.4 混合對照品溶液、供試品溶液、陰性對照溶液的衍生化溶液的制備 分別精密量取“2.2.2”“2.2.3”“2.1.4”項下混合對照品溶液、供試品溶液、陰性對照溶液各5 mL,加1 mol/L異硫氰酸苯酯(PITC)乙腈溶液2.5 mL,然后再加1 mol/L三乙胺乙腈溶液2.5 mL,搖勻,室溫放置1 h,加50%乙腈定容至25 mL,搖勻。量取上述溶液10 mL,置于分液漏斗中,加正己烷10 mL,振搖后靜置分層,取下層溶液,濾過,即得。
2.2.5 專屬性試驗 取“2.2.4”項下混合對照品溶液、供試品溶液、陰性對照溶液的衍生化溶液各適量,按“2.2.1”項下色譜條件進樣測定,記錄色譜,詳見圖3。結果表明,陰性對照在相應位置未見干擾峰,方法專屬性良好。
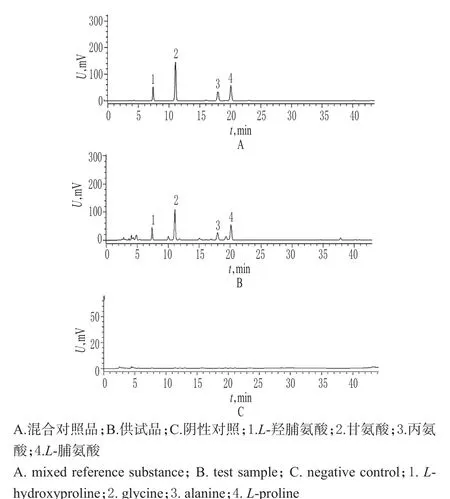
圖3 高效液相色譜圖Fig 3 HPLC chromatograms
2.2.6 線性關系考察 取待測水解氨基酸對照品適量,加0.1 mol/L鹽酸溶液制成L-羥脯氨酸、甘氨酸、丙氨酸和L-脯氨酸質量濃度分別為0.081 9、0.186 2、0.070 3、0.124 2 mg/mL的混合對照品溶液。分別精密量取上述混合對照品溶液1、2、4、6、8 μL,按“2.2.1”項下色譜條件進樣測定,記錄峰面積。以待測水解氨基酸進樣量(x,μg)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得L-羥脯氨酸、甘氨酸、丙氨酸和L-脯氨酸回歸方程,分別為y=1 356 820.6x—254.3(r=0.999 9)、y=2 065 568.1x—7 326.1(r=0.999 8)、y=1 675 750.1x—1 011.6(r=0.999 9)、y=1 561 586.887 8x+65.615 9(r=0.999 9)。結果表明,L-羥脯氨酸、甘氨酸、丙氨酸和L-脯氨酸檢測進樣量線性范圍分別為0.081 9~0.655 2、0.186 2~1.489 6、0.070 3~0.562 0、0.124 2~0.993 6 μg。
2.2.7 精密度試驗 取“2.2.4”項下混合對照品溶液的衍生化溶液適量,按“2.2.1”項下色譜條件連續進樣測定6次,記錄峰面積。結果,L-羥脯氨酸、甘氨酸、丙氨酸和L-脯氨酸峰面積的RSD分別為0.35%、0.38%、0.37%和0.52%(n=6),表明儀器精密度良好。
2.2.8 穩定性試驗 取“2.2.4”項下供試品溶液(批號:1301001)的衍生化溶液適量,分別于室溫下放置0、5、10、15、20、25 h時按“2.2.1”項下色譜條件進樣測定,記錄峰面積。結果,L-羥脯氨酸、甘氨酸、丙氨酸和L-脯氨酸峰面積的RSD分別為1.27%、1.25%、1.48%和1.18%(n=6),表明供試品溶液在室溫放置25 h內基本穩定。
2.2.9 重復性試驗 精密稱取同一批樣品(批號:1301001)適量,按“2.2.3”項下方法制備供試品溶液,按“2.2.4”項下方法制備供試品溶液的衍生化溶液,共6份,再按“2.2.1”項下色譜條件進樣測定,計算含量。結果,L-羥脯氨酸、甘氨酸、丙氨酸和L-脯氨酸平均含量分別為 8.33、16.37、6.96、13.89 mg/g,RSD分別為1.61%、1.94%、1.19%、1.69%(n=6),表明本方法重復性良好。
2.2.10 加樣回收率試驗 取已知含量樣品(批號:1301001)適量,共6份,分別加入一定質量的待測水解氨基酸對照品,按“2.2.3”項下方法制備供試品溶液,另按“2.2.4”項下方法制備供試品溶液的衍生化溶液,再按“2.2.1”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率,結果見表1。

表1 加樣回收率試驗結果(n=6)Tab 1 Results of recovery tests(n=6)
2.2.11 樣品含量測定 取3批樣品各適量,按“2.2.3”項下方法制備供試品溶液,另按“2.2.4”項下方法制備供試品溶液的衍生化溶液,再按“2.2.1”項下色譜條件進樣測定,記錄峰面積并計算樣品含量,結果見表2。結合阿膠三寶膏中阿膠的處方量和制成總量計算4種水解氨基酸的理論含量分別為7.2、16.2、6.3和9.0 mg/g。考慮水分、制劑工藝等影響因素,并結合實際測定結果,暫定限度為每1 g含阿膠以L-羥脯氨酸計,不得少于6.1 mg;以甘氨酸計,不得少于13.8 mg;以丙氨酸計,不得少于5.4 mg;以L-脯氨酸計,不得少于7.7 mg。
3 討論
阿膠是馬科動物驢Equus asinus L.的干燥皮或鮮皮經煎煮、濃縮制成的固體膠,是我國的傳統中藥,具有滋陰補血、潤燥、止血之效。處方中含有阿膠的中藥制劑品種繁多,但由于現行方法對制劑中阿膠的質量控制缺乏針對性,專屬性較差。不法廠家用其他劣質皮類替代驢皮熬制阿膠的違法行為時有發生,導致此類制劑質量良莠不齊,藥效無法得到保障。
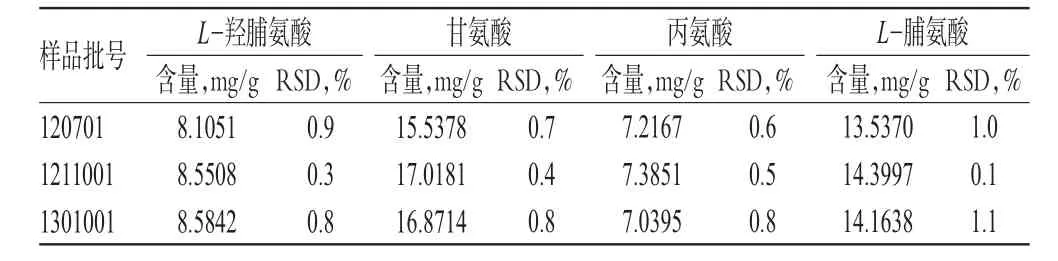
表2 樣品含量測定結果(n=3)Tab 2 Results of contents determination of samples(n=3)
本試驗采用UPLC-MS法,建立了阿膠特征肽的專屬性鑒別,結果表明,本鑒別方法高效、快速、專屬性強,耐用性強,可有效地對阿膠三寶膏中的阿膠進行鑒別。
本試驗參照2015年版《中國藥典》(一部)阿膠項下含量測定的方法,建立了阿膠三寶膏中阿膠4種水解氨基酸的HPLC含量測定項。由于制劑處方中還含有大棗和黃芪,水溶性雜質較多,因此筆者根據制劑的實際情況優化液相條件,延長洗脫時間,以獲得更好的分離度。按優化后的色譜方法進行試驗,所得數據線性關系、精密度、重復性和回收率均良好。
綜上所述,提高的標準能更加有效地控制阿膠三寶膏的質量。
[1] 國家藥典委員會.中華人民共和國藥典:一部[S].2015年版.北京:中國醫藥科技出版社,2015:1010.
[2] 國家藥典委員會.衛生部藥品標準:中藥成方制劑:第二冊[S].WS3-B-0297-90.
[3] 程顯隆.膠類藥材質量控制關鍵技術研究[D].北京:北京中醫藥大學,2014.
[4] 陳再潔,殷金龍,李坤,等.柱前衍生RP-HPLC法測定人參中17種氨基酸的含量[J].中國藥房,2012,23(35):3334-3337.
Study on the Improvement of Quality Standard for Ejiao Sanbao Cream
JIAO Yang,WANG Bing,LIN Yongqiang,XU Lihua(Shandong Institute for Food and Drug Control,Jinan 250101,China)
OBJECTIVE:To improve the quality standard of Ejiao sanbao cream.METHODS:UPLC-MS was adopted to identify the donkey-hide glue in the preparation,the chromatographic conditions:column was Acquity UPLC BEH-C18with mobile phase of acetonitrile-0.1%formic acid(gradient elution),volumetric flow rate was 0.3 mL/min,injection volume was 5 μL,ion source is electrospray ionization source,capillary voltage was 3.5 kV,capillary outlet voltage was 120 V,cone voltage was 50 V,desolvation temperature was 400℃,drying gas flow was 10 L/min,nebulizer pressure was 40 psi,collision energy was 10-45 V,scan range was m/z 200-1 500;detection mode was positive ion mode(ESI+),multiple reaction monitoring.HPLC was used to determine the contents of 4 hydrohyzing amino acid:column was Agela Venusil XBP-C18with mobile phase A of acetonitrile-0.1 mol/ L sodium acetate solution(36%acetic acid adjusted to pH 6.5)(7∶93,V/V)and B of 80%acetonitrile solution(gradient elution)at a flow rate of 1.0 mL/min,the detection wavelength was 254 nm,and column temperature was 43℃.RESULTS:The UPLC-MS peak of Equus asinus was clear and specific,without negative interference.The linear range was 0.081 9-0.655 2 μg for L-hydroxyproline(r=0.999 9),0.186 2-1.489 6 μg for glycine(r=0.999 8),0.070 3-0.562 0 μg for alanine(r=0.999 9)and 0.124 2-0.993 6 μg for L-proline(r=0.999 9);RSDs of precision,stability and reproducibility tests were lower than 2.0%;recoveries were 99.85%-103.69%(RSD=1.35%,n=6),99.91%-103.93%(RSD=1.46%,n=6),96.86%-101.27%(RSD=1.69%,n=6)and 97.44%-101.45%(RSD=1.54%,n=6),respectively.CONCLUSIONS:The improved standard can more effectively control the quality of Ejiao sanbao cream.
Ejiao sanbao cream;Quality standard;UPLC-MS;HPLC
R927.2
A
1001-0408(2017)03-0376-05
2016-02-09
2016-04-25)
(編輯:張 靜)
國家科技重大專項課題(No.2014ZX09304307-002-002);山東省自然科學基金資助項目(No.ZR2013HM074)
*藥師。研究方向:藥物分析。電話:0531-81216522。E-mail:jiaoyang579@hotmail.com
DOI10.6039/j.issn.1001-0408.2017.03.25
關鍵詞阿膠三寶膏;質量標準;超高效液相色譜-質譜聯用法;高效液相色譜法
