中國節節麥基于ISSR標記的遺傳多樣性分析
2017-03-01 07:59:31李玉閣蘇亞中張大樂李鎖平
麥類作物學報 2017年1期
李玉閣,蘇亞中,張大樂,李鎖平
(河南大學生命科學學院,河南開封 475004)
中國節節麥基于ISSR標記的遺傳多樣性分析
李玉閣,蘇亞中,張大樂,李鎖平
(河南大學生命科學學院,河南開封 475004)
為揭示中國節節麥的遺傳多樣性并發掘可能具有獨特變異的節節麥種質資源,采用ISSR標記對75份中國節節麥的遺傳多樣性進行分析。結果表明,篩選出的9條ISSR引物共檢測得到155個位點,多態性位點(138個)百分率為89.31%。Nei’s多樣性指數(He)、Shannon’s信息指數(I)、基因分化系數(Gst)和基因流(Nm)分別為0.273 4、0.415 2、0.138 5和3.11,表明中國節節麥有較高的遺傳多樣性,群體間有中等程度的遺傳分化。基于簡單相似系數的UPGMA聚類分析表明,除5份河南和陜西節節麥聚類形成獨立的分支Group 2 (T078、T102和SC1) 和Group 3(SX38和T006)外,絕大多數節節麥均聚類形成較大的分支Group 1,且在Group 1中,除4份節節麥(T002、T023、XJ6和XJ49)外,絕大多數節節麥均依據地理分布分別形成了黃河流域節節麥亞組和新疆節節麥亞組,在遺傳距離約0.77處,又可依次分為河南、陜西和新疆節節麥小分支。二維主成分分析、群體的遺傳一致度和分子變異分析也表明了類似的結果,同屬于黃河流域節節麥亞組的河南、陜西節節麥遺傳一致度(S=0.971 8)較高,群體內個體差異是中國節節麥變異的主要原因,但兩大亞組和三居群的遺傳差異分別占總變異的18.57%和10.38%。可見,不同節節麥的地理分布和生境差異,是導致中國節節麥居群遺傳分化的主要原因,而聚類分析中單獨形成獨立分支Group 2和Group 3的5份黃河流域節節麥,可能是具有獨特遺傳變異的種質資源。
中國節節麥;ISSR;聚類分析;二維主成分分析;分子變異分析
自然遺傳變異是植物遺傳改良重要的基因資源。節節麥(Aegilopstauschii,DD,2n=14)作為普通小麥(TiriticumaestivumL.,AABBDD,2n=42)D基因組的祖先供體種,不僅具有比普通小麥D基因組更豐富的遺傳變異,而且其寶貴基因資源易于通過傳統雜交導入普通小麥,因此,節節麥被看作是小麥族中可應用于小麥遺傳改良最重要的遺傳資源[1-5]。Mizuno等[6]和Sohail等[7]對世界不同地理分布的節節麥種群遺傳結構的分析表明,少數隸屬于L2譜系2-3亞系的節節麥,至少是L2譜系的節節麥,主要參與了普通小麥D基因組的起源,而具有更廣泛遺傳變異、隸屬于L1譜系的節節麥,沒有參與普通小麥的起源,是小麥遺傳改良重要而豐富的遺傳資源。分布于我國新疆西部的伊犁河谷和黃河流域的節節麥,隸屬于L1譜系的節節麥,不僅沒有參與普通小麥的起源,且部分來源于河南、陜西的黃河流域節節麥,還具有其他譜系節節麥所缺乏的獨特遺傳變異[6]。因此,系統采集中國節節麥,對其進行廣泛的遺傳多樣性分析,并從中篩選具有獨特遺傳變異的種質資源,無論對節節麥的遺傳多樣性保護,還是對小麥的遺傳改良來講均具有重要的意義。
2006-2008年本課題組對分布于我國河南、陜西、新疆的節節麥野生種質資源進行了系統調查和采集,并相繼對中國黃河流域[8]和新疆伊犁地區[9]節節麥的SSR遺傳多樣性、HMW-GS組成[10-11]、穗發芽[12]和抗旱特性[13]進行了大規模的初步調查和比較分析。為進一步揭示中國節節麥的遺傳多樣性和居群間的遺傳差異,并從中篩選出具有獨特變異的寶貴種質資源,本研究在黃河流域和新疆節節麥SSR遺傳多樣性分析的基礎上,選取有代表性的75份節節麥,采用能很好揭示物種內遺傳多樣性、且有效性已在伊朗節節麥遺傳多樣性分析中得以驗證的ISSR分子標記[11-17],進行遺傳多樣性分析。
1 材料與方法
1.1 供試材料
75份代表性中國節節麥(Aegilopstauchii)種質資源(表1),由河南大學生命科學學院植物種質資源與遺傳實驗室收集并保存。
1.2 試驗方法
1.2.1 基因組DNA的提取
隨機選取20粒左右的節節麥種子,在溫室中培養5~7 d至黃化苗,采用CTAB法從嫩葉中提取基因組DNA。
1.2.2 ISSR引物的選擇、合成與篩選
首先,選擇Baranduzi等[14]、杜金昆等[16]和馬艷明等[17]研究中得到的擴增效果良好的18條ISSR引物,交由上海生物工程科技有限公司合成。然后,根據遺傳多樣性的SSR分析結果[8-9],選取遺傳差異相對較大的部分節節麥為材料,對18條ISSR引物作進一步篩選,將擴增多態性好、重復性強的引物作為本研究用于遺傳多樣性分析的引物。
1.2.3 PCR擴增及結果檢測
PCR擴增采用15 μL的反應體系,包括約50 ng的基因組DNA、100 μmol·L-1的dNTPs、0.5 U的TaqDNA聚合酶(TaKaRa)和0.5 μmol·L-1的ISSR引物。PCR循環程序:94 ℃預變性4 min;94 ℃變性30 s,50~60 ℃(根據引物的Tm值調節退火溫度,一般為Tm+2 ℃)退火45 s,72 ℃延伸60 s,32個循環;72 ℃延伸15 min。PCR擴增產物采用非變性聚丙烯酰胺凝膠電泳和銀染法分別進行分離和鑒定,具體方法參照蘇亞蕊等[7]。
1.2.4 ISSR分析結果的統計與分析
每條ISSR引物至少PCR擴增兩次,只有那些能穩定重復的清晰條帶才作為可能的多態性條帶進行統計。在每個凝膠上,根據對應的ISSR擴增條帶的有無記錄條帶,有擴增條帶的記為1,沒有擴增條帶的記為0,將擴增結果轉換成二元矩陣。采用NTsys2.1軟件,基于簡單匹配系數(simple matching coefficient,SM),對75份中國節節麥進行UPGMA(un-weighted pair group method analysis)聚類分析和二維主坐標分析(principal coordinates analysis,PCoA)。應用POPGENE.32軟件計算河南、陜西和新疆節節麥居群的觀測等位基因數(observed number of alleles,Na)、有效等位基因數(effective number of alleles,Ne)、Nei’s基因多樣性指數(Nei’s gene diversity,He)、Shannon’s信息指數(Shannon’s information index,I)、多態位點百分率(percentage of polymorphic loci,PPB)、不同居群間的總基因多樣性(total gene diversity,Ht)、群體內的基因多樣性(gene diversity within population,Hs)、群體間的基因多樣性(gene diversity among populations,Dst)、遺傳分化系數(genetic differentiation coefficient,Gst)、基因流(gene flow,Nm)、Nei’s遺傳距離(genetic distance,D)和遺傳一致度(genetic identity,S),并依據遺傳距離進行群體聚類和分組。應用DCFA軟件對數據進行轉化,然后用AMOVA 1.55軟件進一步對中國節節麥進行分子變異分析。

表1 75份代表性中國節節麥材料的編號及來源Table 1 Name and origin of the 75 Chinese Aegilops tauchii accessions
2 結果與分析
2.1 ISSR-PCR擴增結果
利用篩選出的9條ISSR引物,從75份中國節節麥中共檢測得到155條清晰、可重復、片段大小在120~2 000 bp的PCR條帶,每條引物可擴增得到條帶12~24條,多態性條帶在9~21條,多態性位點百分率為89.31%(表2)。從引物UBC859的擴增結果(圖1)可以初步看出,新疆伊犁地區節節麥有較高的遺傳相似性,而來自河南、陜西的黃河流域節節麥,有較為廣泛的遺傳變異。

表2 篩選出的9條ISSR引物及其擴增結果Table 2 Nine selected primers and their amplified resluts
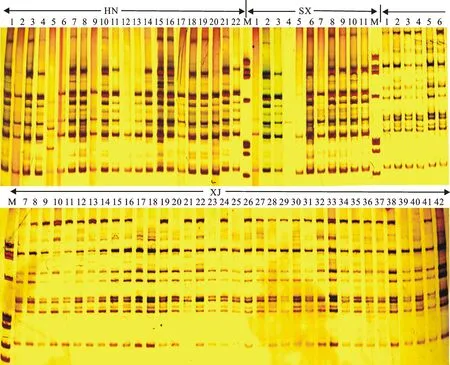
HN、SX和XJ分別代表河南、陜西和新疆的節節麥材料;M代表X174-HaeⅢ digest DNA marker。 下同。
HN,SX and XJ represent the materials from Henan,Shaanxi and Xinjiang provinces,respectively; M represents the X174-HaeⅢ digest DNA marker.The same below.
圖1 引物UBC859的PCR擴增結果
Fig.1 Profiles of PCR products amplified with the primer UBC859
2.2 75份中國節節麥ISSR遺傳多樣性的UPGMA聚類分析和二維主成分分析
為進一步揭示75份中國節節麥的親緣關系,基于簡單相似系數,利用NTsys2.1對ISSR遺傳多樣性結果進行聚類分析和二維主成分分析。從聚類結果(圖2)可以看出,在相似系數約0.67處,除3份來自河南(T078、T102和T006)和2份來自陜西(SC1和SX38)的節節麥分別形成相對獨立的分支Group 2和Group 3外,其他70份中國節節麥形成較大的分支Group 1。而且,除T002、T023、XJ6和XJ49(虛線框表示)這4份材料外,在相似系數約0.71處,Group 1中來自新疆伊犁地區的節節麥(Sub 1)和來自河南、陜西的黃河流域節節麥(Sub 2)又形成了2個不同的亞組。而在相似系數約0.77處,分布在黃河流域節節麥亞組Sub 2的節節麥,又能明確地分為河南、陜西和新疆節節麥小分支。對遺傳距離與系統進化樹之間的相關性分析表明,二者存在極顯著的相關性(r=0.812 6,P<0.01)。對75份節節麥的二維主成分分析表明(圖3),主要代表新疆和黃河流域節節麥地理差異的第1、2主坐標的方差貢獻率分別為51.41%和6.24%。可見,聚類分析與主成分分析結果基本一致,均表明地理隔離是造成黃河流域節節麥和新疆伊犁地區節節麥遺傳差異的主要原因。而且,從75份節節麥的分布可以直觀看出,新疆節節麥的遺傳相似度較高,而來自河南、陜西的黃河流域節節麥遺傳多樣性相對豐富,尤其是形成獨立分支Group 2和Group 3的5份節節麥,可能是具有獨特遺傳變異的節節麥種質資源。

圖2 75份中國節節麥ISSR遺傳多樣性分析的系統聚類圖

圖3 75份中國節節麥的二維主坐標分析
2.3 中國節節麥居群的遺傳多樣性和遺傳分化程度分析
為進一步揭示中國節節麥群體及不同居群間的遺傳多樣性水平和遺傳分化程度,采用POPGENE.32軟件對河南、陜西和新疆節節麥居群的群體基因遺傳多樣性(表3)、群體基因的遺傳分化程度(表4)、三居群間的遺傳一致度和遺傳距離(表5)參數進行計算和比較分析,并依據遺傳距離對不同居群進行聚類分析(圖4)。結果表明,陜西節節麥的群體遺傳多樣性水平(He=0.242 0,I=0.366 0)略高于新疆(He=0.239 9,I=0.360 3)和河南(He=0.228 1,I=0.354 7)。新疆節節麥的多態性位點比率最低,僅為71.04%。中國節節麥群體的總基因多樣性為0.274 7,不同群體間發生一定的遺傳分化(Gst=13.85%),并存在中等程度的基因交流(Nm=3.11)。基于遺傳距離對不同居群節節麥的UPGMA聚類結果也表明,中國節節麥可分為黃淮流域節節麥和新疆伊犁地區節節麥兩大居群,而河南節節麥和陜西節節麥有較高的遺傳一致度(S=0.971 8),遺傳距離較近(D=0.039 7)。采用AMOVA 1.55軟件對黃河流域節節麥和新疆伊犁地區節節麥(Groups)及河南、陜西和新疆節節麥(populations)的分子變異分析(表6)也進一步表明了類似的情況,黃河流域與新疆伊犁地區節節麥的遺傳變異占總變異的18.57%,而河南、陜西、新疆節節麥的遺傳變異占總變異的10.38%。群體間存在較明顯的遺傳分化。

表3 不同居群的中國節節麥的遺傳多樣性分析Table 3 Genetic diversity of three populations of Chinese Aegilops tauchii accessions

表4 中國節節麥群體基因遺傳多樣性的Nei’s分析Table 4 Nei’s analysis of gene diversity of Chinese Aegilops tauchii accessions
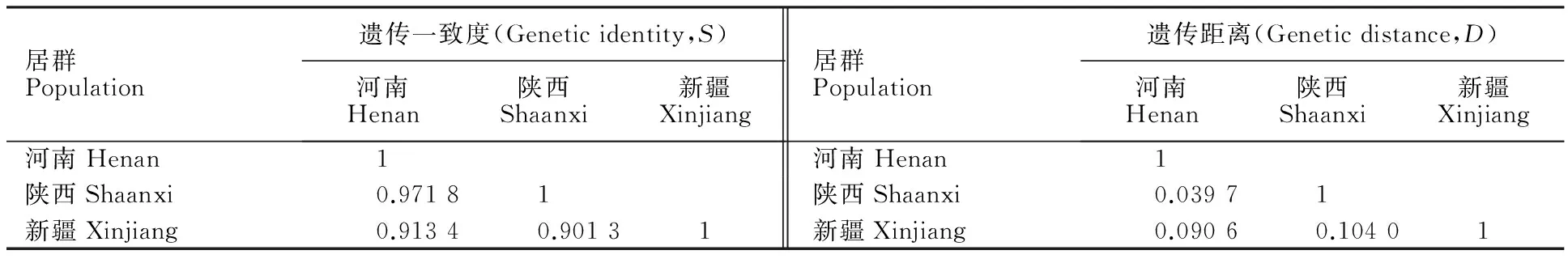
表5 三個中國節節麥居群的遺傳一致性和遺傳距離分析Table 5 Genetic distance and similarity among three populations of Chinese Aegilops tauchii accessions
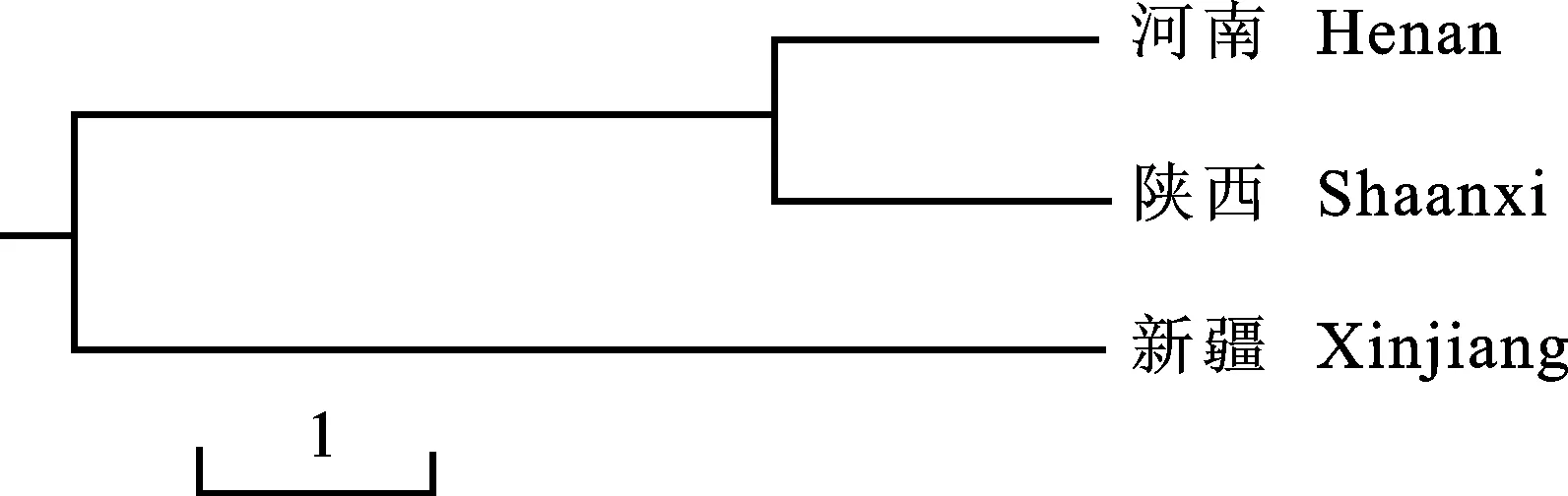
圖4 河南、陜西和新疆節節麥居群的UPGMA遺傳距離聚類圖
3 討 論
具有廣泛的地理分布和自然變異的節節麥(Aegilopstauschii),是小麥族野生物種中最重要也最易于用于小麥遺傳改良的寶貴種質資源[3-4]。而在我國黃淮麥區有良好適應性的黃河流域節節麥,不僅沒有參與普通小麥的起源,隸屬于存在更廣泛遺傳變異的L1居群,且部分來源于河南、陜西的黃河流域節節麥,還具有其他節節麥所缺乏的獨特遺傳變異[6-7]。因此,系統采集并開展中國節節麥的遺傳多樣性分析,從中篩選具有獨特遺傳變異的種質資源,無論對節節麥的物種多樣性保護,節節麥的起源和傳播研究,還是對中國節節麥在小麥遺傳改良中的有效利用,均具有重要的理論價值和現實意義。
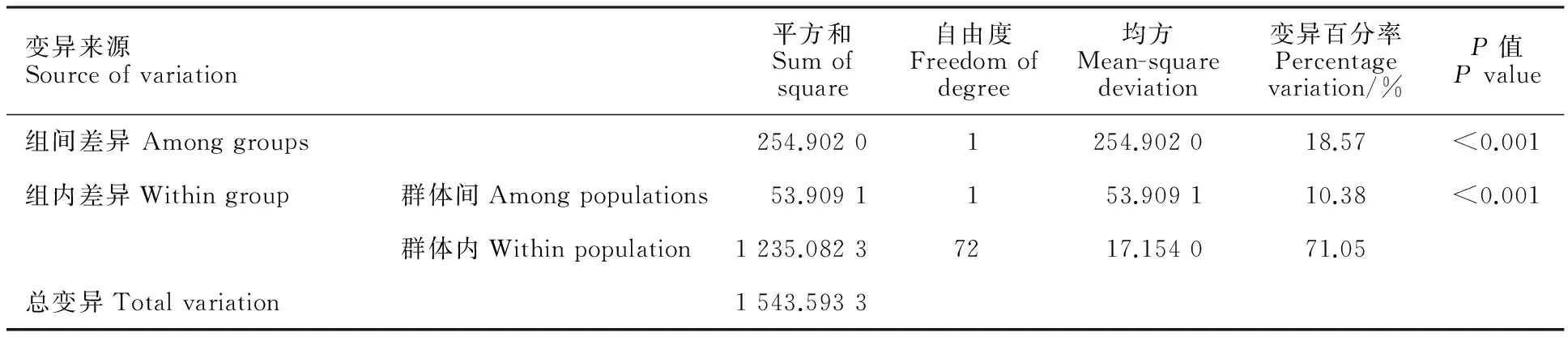
表6 中國節節麥群體的分子變異分析Table 6 Analysis of molecular variation of Chinese Aegilops tauchii accessions
目前對節節麥的遺傳多樣性分析雖已有不少報道[7-8,18-22],但對中國節節麥遺傳多樣性和群體間分化程度的比較分析卻并不多[20]。本課題組基于中國節節麥對小麥遺傳改良中的重要性,對其進行了系統的采集和大規模的遺傳多樣性分析和相關優良性狀的初步鑒定和分析[8-13],本研究主要在已開展的黃河流域[8]和新疆伊犁地區節節麥[9]SSR遺傳多樣性分析的基礎上,從中篩選出75份有代表性的中國節節麥,進一步開展遺傳多樣性的ISSR分析,并期望通過群體間的遺傳分化程度比較和聚類分析,從中篩選出可能具有獨特遺傳變異的種質資源。研究結果與SSR[8-9,20]的分析基本一致,黃河流域節節麥和新疆伊犁節節麥有較顯著的遺傳差異。除個別例外節節麥外,絕大多數中國節節麥,可依據其地理分布進行分組,其中,存在地理隔離的新疆伊犁地區節節麥和黃河流域節節麥的差異較大,河南和陜西節節麥的遺傳一致度較高,不同群體間存在較為明顯的遺傳分化(PHist=0.289,Gst=0.138 5)。按照Buso等[23]的分類,屬于中等程度的遺傳分化。而三群體間的基因流為3.11>1,表明三群體間的遺傳分化可能與遺傳漂變無關。根據NTsys、POPGENE和AMOVA分析結果均可以看出,不同節節麥的地理分布和生境差異,可能是造成中國不同居群節節麥差異的主要原因。
此外,與Mizuno等[6]和Sohail等[7]對世界不同地理分布的節節麥的種群遺傳結構分析類似,本研究也表明,在黃淮麥區有良好適應的黃河流域節節麥,部分具有獨特的遺傳變異。在分析的75份中國節節麥,盡管絕大多數在遺傳距離約0.71處均聚類形成一個較大的分支Group 1,但有5份來源于河南(T078、T102和T006)和陜西(SC1和SX38)的節節麥,卻在聚類分析和二維主成分分析中形成了兩個明顯獨立的分支Group 2和Group 3。實驗室前期對黃河流域[11]和新疆伊犁地區[9]節節麥HMW-GS的組分分析也表明,盡管新疆伊犁地區節節麥的HMW-GS組成比較一致(均為2+10亞基組合類型),黃河流域節節麥也以5t+10.5t亞基組合為主要的組合類型(分布頻率高達96%),但ISSR分析中位于Group 3的節節麥T006,其HMW-GS組卻為分布頻率較低的6.2t+10.4t組合類型。李玉閣[24]對部分中國節節麥LMW-GS和α-、γ-醇溶蛋白的基因克隆和分子特征分析也表明,T006和SC1的LMW-GS基因與對品質可能有較大貢獻的D3-2和D3-4單倍型有較高的同源性,其α-、γ-醇溶蛋白基因的分子特征上存在獨特的遺傳變異。其α-醇溶蛋白基因部分與A、B基因組來源的α-醇溶蛋白基因有更高的同源性,不含或含有較少的、可誘發人類最常見的一種慢性遺傳性疾病-乳糜瀉(celiac diesease,CD)的免疫優勢多肽,其絕大部分γ-醇溶蛋白也與B基因組來源的、存在于強筋優質小麥的基因有更高的同源性,在N-末端重復區含有一個額外的半胱氨酸殘基。因此,綜合以上分析結果,本研究推測這5份節節麥,至少是實驗已開展分析的T006和SC1,可能是節節麥遺傳多樣性保護和小麥遺傳改良可利用的獨特種質資源。對其LMW-GS和α-、γ-醇溶蛋白的基因獨特分子特征及其在小麥品質改良和CD預防中的應用價值的進一步深入分析和鑒定,目前正在的試驗中。
有關中國黃河流域節節麥和新疆節節麥的傳播關系,目前尚存在一定爭議。有研究[8,19-20,25-26]認為黃河流域節節麥可能是作為普通小麥的伴生種,從伊朗經絲綢之路傳入我國的,而新疆伊犁地區節節麥是中東節節麥野生居群在東部的延續,二者不存在直接的傳播關系。也有研究[27]認為,中國節節麥至少起源于兩個不同的居群,新疆節節麥可能起源于黃河流域節節麥,但也不排除黃河流域節節麥是從新疆傳播而來的可能。而本研究中對中國節節麥的ISSR遺傳多樣性的聚類分析也發現,在系統進化樹中有兩份河南節節麥(T002和T023)聚類在了新疆亞組,而兩份新疆節節麥(XJ6和XJ49)卻聚類在了黃河流域節節麥亞組,與陜西節節麥有更高的同源性。可見,對代表性中國節節麥、以及在聚類分析中處于特殊地位的節節麥與國外節節麥,采用更多的分子標記類型、進行更廣泛的遺傳多樣性分析,也有望為揭示黃河流域節節麥和新疆節節麥的傳播關系研究,提供更多的證據和參考。
[1]DVORAK J,LUO M C,YANG Z L,etal.The structure of theAegilopstauschiigenepool and the evolution of hexaploid wheat [J].TheoreticalandAppliedGenetics,1998,97(4):657.
[2]REIF J C,ZHANG P,DREISIGACKER S,etal.Wheat genetic diversity trends during domestication and breeding [J].TheoreticalandAppliedGenetics,2005,110(5):859.
[3]NAGHAVI M R,MARDI M.Characterization of genetic variation among accessions ofAegilopstauschii[C].Proceedings Asia Pacific Conference on Plant Tissue and Agribiotechnology,2010,18(1):93-96.
[4]SOHAIL Q,INOUE T,TANAKA H,etal.Applicability ofAegilopstauschiidrought tolerance traits to breeding of hexaploid wheat [J].BreedingScience,2011,61(4):347.
[5]SINGH S,CHAHAL G S,SINGH P K,etal.Discovery of desirable genes in the germplasm pool ofAegilopstauschiiCoss.[J].IndianJournalofGeneticsandPlantBreeding,2012,72(3):271.
[6]MIZUNO N,YAMASAKI M,MATSUOKA Y,etal.Population structure of wild wheat D-genome progenitorAegilopstauschiiCoss.: implications for intraspecific lineage diversification and evolution of common wheat [J].MolecularEcology,2010,19(5):999.
[7]SOHAIL Q,SHEHZAD T,KILIAN A,etal.Development of diversity array technology (DArT) markers for assessment of population structure and diversity inAegilopstauschii[J].BreedingScience,2012,62(1):38.
[8] 蘇亞蕊,張大樂,徐守明,等.粗山羊草居群間遺傳分化及多樣性的SSR分析[J].生態學報,2010,30(4):969.
SU Y R,ZHANG D L,XU S M,etal.Genetic diversity and differentiation in differentAegilopstauschiipopulations revealed by SSR [J].ActaEcologicaSinica,2010,30(4):969.
[9] 賀松濤.新疆節節麥遺傳多樣性分析及耐鹽材料的篩選[D].開封:河南大學,2010:29.
HE S T.Analysis of genetic diversity forAegilopstauschiiin Xinjiang and identifition for salt-tolerance [D].Kaifeng: Henan University,2010:29.
[10] 蘇亞蕊,張大樂,張 明,等.黃河中游粗山羊草三種y-型高分子量谷蛋白亞基的鑒定、克隆及系統進化分析[J].作物學報,2009,35(7):1244.
SU Y R,ZHANG D L,ZHANG M,etal.Characterization,molecular cloning and phylogenetic analysis of three y-type high molecular weight glutenin subunit genes fromAegilopstauschiiof the Middle Reaches of Yellow River [J].ActaAgonomicaSinica,2009,35(7):1244.
[11] 張 明,蘇亞蕊,張大樂,等.黃河中游地區粗山羊草高分子量谷蛋白亞基組成分析[J].華北農學報,2008,23(3):28.
ZHANG M,SU Y R,ZHANG D L,etal.The analyse of allelic composition of the HMW glutenin subunits inAegilopstauschiiin Middle Reaches of the Yellow River [J].ActaAgriculturaeBoreali-Sinica,2008,23(3):28.
[12] 蘇亞蕊,劉新浩,張大樂,等.黃河中游地區節節麥抗穗發芽的鑒定與分析[J].植物遺傳資源學報,2011,12(6):1004.
SU Y R,LIU X H,ZHANG D L,etal.Screening and analysis of pre-harvest sprouting resistant germplasm fromAegilopstauschiis in the Middle Reaches of the Yellow River [J].JournalofPlantGeneticResources,2011,12(6):1004.
[13] 方 圓.節節麥抗旱性種質資源篩選及其轉錄組分析[D].開封: 河南大學,2014:61.
FANG Y.The drought resistant germplasm screening and transcriptome analysis ofAegilopstauschii[D].Kaifeng: Henan University,2014:61.
[14]BARANDUZI J A,SOFALIAN O,ZAKARIA A R,etal.Assessment of genetic diversity inAegilopsspecies in North-West of Iran using ISSR marker [J].YüzüncüYlUniversityJournalofAgriculturalSciences,2013,2:66.
[15]DASHCHI S,MANDOULAKANI B A,DARVISHZADE R,etal.Molecular similarity relationships among Iranian bread wheat cultivars and breeding lines using ISSR markers [J].NotulaeBotanicaeHortiAgrobotaniciCluj-Napoca,2012,40(2):254.
[16] 杜金昆,姚穎垠,倪中福,等.普通小麥、斯卑爾脫小麥、密穗小麥和輪回選擇后代材料ISSR分子標記遺傳差異研究[J].遺傳學報,2002,29(5):445.
DU J K,YAO Y Y,NI Z F,etal.Genetic diversity revealed by ISSR molecular marker in common wheat spelt,compactum and progeny of recurrent selection [J].ActaGeneticaSinica,2002,29(5):445.
[17] 馬艷明,李斯深,范玉頂,等.黃淮麥區小麥品種的ISSR位點遺傳多樣性分析[J].植物遺傳資源學報,2006,7(1):13.
MA Y M,LI S S,FAN Y D,etal.Genetic diversity of ISSR loci for wheat cultivars of Huang-Huai winter wheat region [J].JournalofPlantGeneticResources,2006,7(1):13.
[18]JAASKA V.Aspartate aminotransferase and alcohol dehydrogenase isoenzymes: Intraspecific differentiation inAegilopstauschiiand the origin of the D genome polyploids in the wheat group [J].PlantSystematicsandEvolution,1981,137(4):259.
[19]PESTSOVA E,GANAL M W,RODER M S.Isolation and mapping of microsatellite markers specific for the D genome of bread wheat [J].Genome,2000,43(4):689.
[20]WEI H T,LI J,PENG Z S,etal.Relationships ofAegilopstauschiirevealed by DNA fingerprints: The evidence for agriculture exchange between China and the West [J].ProgressinNaturalScience,2008,18:1525.
[21] 王亞娟,王長有,劉新倫,等.基于SSR標記的粗山羊草遺傳多樣性分析 [J].農業生物技術學報,2010,18(3):493.
WANG Y J,WANG C Y,LIU X L,etal.Genetic diversity ofAegliopstauschiibased on SSR marker [J].JournalofAgriculturalBiotechnology,2010,18(3):493.
[22] 蘭秀錦,魏育明,王志容,等.中國節節麥與中東節節麥的醇溶蛋白遺傳多樣性比較研究 [J].四川農業大學學報,1999,17(4):245.
LAN X J,WEI Y M,WANG Z R,etal.Gliadin comparison betweenAegilopstauschiiCosson from China and Middle-East [J].JournalofSichuanAgriculturalUniversity,1999,17(4):245.
[23]BISO G S C,RANGEL P H,FERREIRA M E.Analysis of genetic variability in South American wild rice populations (Oryzaglumaepatula) with isozymes and RAPD markers [J].MolecularEcology,2002,7(1):107.
[24] 李玉閣.普通小麥和中國節節麥LMW-GS和α-,γ-醇溶蛋白基因的克隆與序列分析[D].開封:河南大學,2014:135.
LI Y G.Molecular cloning and sequence analysis of LMW-GS,α-,γ-gliadin genes from common wheat andAegilopstauschiinative to China [D].Kaifeng:Henan University,2014:135.
[25] 陳文杰,劉登才.基于高分子谷蛋白基因的中國節節麥系統關系[EB/OL].北京:中國科技論文在線,(2012-12-19)[2016-04-01].http:// www.paper.edu.cn /releasepaper /content/201212-489.
CHEN W J,LIU D C.Phylogenetic relationship among ChineseAegilopstauschiiaccessions based on high-molecular-weight glutenin genes [EB/OL].Beijing:Sciencepaper online,(2012-12-19)[2016-04-01].http:// www.paper.edu.cn / releasepaper /content/201212-489.
[26] 顏 濟,楊俊良,崔乃然,等.新疆伊犁地區的節節麥 (AegilopstauschiiCosson)[J].作物學報,1984,10(1):1.
YAN J,YANG J L,CUI N R,etal.AegilopstauschiiCosson in Yili,Xinjiang [J].ActaAgronomicaSinica,1984,10(1):1.
[27] 王 慶,黃 林,袁中偉,等.中國新疆與黃河流域節節麥的傳播關系[J].四川農業大學學報,2010,28(4):407.
WANG Q,HUANG L,YUAN Z W,etal.Transimission relationship betweenAegilopstauchiiCossons in Xinjiang and Yellow River basin in China [J].JournalofSichuanAgriculturalUniversity,2010,28(4):407.
Genetic Diversity ofAegilopstauchiiAccessions Native to China Revealed by ISSR Markers
LI Yuge,SU Yazhong,ZHANG Dale,LI Suoping
(College of Life Science,Henan University,Kaifeng,Henan 475004,China)
In order to reveal the genetic diversity ofAegilopstauchiiaccessions native to China extensively and screen some beneficial wild resources with individual genetic variation,ISSR markers were used to assess the genetic diversity of 75 ChineseAegilopstauchiiaccessions. Nine ISSR primers produced 138 (89.31%) polymorphic bands out of 155 bands in total. Genetic parameters including average number of effective alleles (Ne=1.471 5),Nei’s gene diversity (He=0.273 4),Shannon’s information index (I=0.415 2),and gene differentiation coefficient (Gst=0.138 5) revealed a high level of genetic diversity and middle level of gene flow maintained in ChineseAelilopstauchiipopulations.A dendrogram constructed on the basis of cluster analysis of similarity simple coefficient clustered all the samples in three main groups 1,2 and 3. Although most of accessions except for T002,T023,XJ6 and XJ49 were placed into group 1 with two subgroups (sub 1 and sub 2),regarding to their different locations (Yellow River basin and Yili River Valley in Xinjiang). There were still five accessions from Henan and Shaanxi provinces clustered in group 2 (T078,T102 and SC1),and group 3 (SX38 and T006). The further two-dimensional principal coordinates analysis (PCoA),the analysis on the genetic distance and similarity among Henan,Shaanxi and Xinjiang populations and the analysis of molecular variance (AMOVA) also confirmed the results that the genetic similarity of Henan and Shaanxi was highest among the three populations. 18.57% of the variance was due to the differences between Yellow River basin and Yili Valley,and 10.38% of the variance was due to differences among populations of Henan,Shaanxi and Xinjiang and the remaining 71.05% was due to differences within populations. Thus,it was suggested that the high genetic diversity of ChineseAegilopstauchiiwas attributable to geographic isolation,and the five accessions constituted two independent groups 2 and 3 maybe the beneficial wild resources with individual genetic variations.
Aegilopstauchiiof China; ISSR; Phylogenic analysis; PCoA; AMOVA
時間:2017-01-03
2016-06-06
2016-12-15
國家自然科學基金面上項目(31271713);河南省高等學校重點科研項目(15A180011)
E-mail:lygzixiu@henu.edu.cn
李鎖平(E-mail:lisuoping@henu.edu.cn)
S512.9;S330
A
1009-1041(2017)01-0030-10
網絡出版地址:http://www.cnki.net/kcms/detail/61.1359.S.20170103.1625.010.html
猜你喜歡
國畫家(2022年2期)2022-04-13 09:07:46
四川文學(2021年4期)2021-07-22 07:11:54
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
絲綢之路(2014年9期)2015-01-22 04:24:46
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
兒童與健康(2011年4期)2011-04-12 00:00:00
新疆人文地理(2009年7期)2009-09-29 09:56:14
