分步預硫化Mo-Ni/γ-Al2O3催化劑的制備、表征及加氫催化性能
2017-02-08 01:37:44王鼎聰趙德智楊辰思張志偉
石油學報(石油加工) 2017年1期
關鍵詞:催化劑
張 強, 丁 巍,3, 王鼎聰, 趙德智, 楊辰思, 張志偉
(1.遼寧石油化工大學 化學化工與環境學部, 遼寧 撫順 113001; 2.中國石化 撫順石油化工研究院, 遼寧 撫順 113001;3.中國石油大學 重質油國家重點實驗室, 北京 102249)
?
分步預硫化Mo-Ni/γ-Al2O3催化劑的制備、表征及加氫催化性能
張 強1, 丁 巍1,3, 王鼎聰2, 趙德智1, 楊辰思1, 張志偉1
(1.遼寧石油化工大學 化學化工與環境學部, 遼寧 撫順 113001; 2.中國石化 撫順石油化工研究院, 遼寧 撫順 113001;3.中國石油大學 重質油國家重點實驗室, 北京 102249)
采用自組裝法將硫脲、檸檬酸及聚乙二醇-200制成器外預硫化沉淀液,原位沉淀法制備了分步預硫化Mo-Ni/γ-Al2O3催化劑。采用BET、XRD、CO原位吸附、XPS、TEM等手段表征所制備的催化劑,采用固定床微型反應器評價催化劑的加氫催化性能,并與直接器內預硫化法制備的催化劑對比。結果表明,分步預硫化法制備的催化劑在第一次器外預硫化后硫化度達到74.8%;聚乙二醇-200和硫脲的加入可以改善催化劑孔性質,且當硫脲含量達到20%(占Mo、Ni金屬氧化物質量和)時,催化劑的比表面積達到198 cm2/g,此時,其活性金屬負載分散性好,硫化度比對比劑提高了2.60百分點。分步預硫化Mo-Ni/γ-Al2O3催化劑催化FCC柴油加氫反應40 h的加氫脫硫、脫氮率及芳烴飽和率比對比劑分別高出2.28百分點、1.94百分點及3.46百分點。
分步預硫化; 原位沉淀; 催化劑; 加氫脫硫(HDS); 加氫脫氮(HDN)
面對現今石油能源環境,重油和渣油加氫技術已經成為主要的研究方向[1-3],催化劑更是其中的重點。載體性質、活性金屬負載方式等直接影響加氫催化劑的活性。載體是催化劑的關鍵組成部分,對催化劑的孔結構、表面酸性、電性質等都有很大影響[4]。活性氧化鋁作為催化劑載體效果顯著[5-6]。王鼎聰[7]采用納米自組裝方法合成了大孔容介孔氧化鋁,田野等[8]以此為載體制備的催化劑在劣質油加氫中表現出了良好的活性及穩定性,鄢景森等[9]研究的TiO2-Al2O3載體表現出了良好的孔結構性質及較高的金屬負載分散性。
加氫催化劑的活性組分Mo、Ni、Co、W等元素一般以金屬氧化物形式存在,在進行加氫反應前要進行預硫化處理,以提高催化劑的活性和選擇性[10-13]。人們對預硫化機理已經進行了廣泛的研究[14],器內預硫化技術已很成熟,器外預硫化技術也有很多研究報道。GAO等[15]詳細研究并比較了器內及器外預硫化技術的特點,夏遠亮[16]采用原位分解法制備了免預硫化CoMo/γ-Al2O3催化劑,林凌等[17]制備的免預硫化加氫脫硫MoNiP/Al2O3催化劑擁有較高的催化活性,田維乾等[18]制備的器外預硫化催化劑表現出較高的催化特性。但器外預硫化技術由于預硫化液黏度大等原因,實現工業化和大規模生產有諸多困難[19]。
筆者將硫脲及聚乙二醇-200直接加入到活性金屬浸漬液中,浸漬后置于微型反應器中進行原位沉淀反應,制備一種免焙燒、分步預硫化的加氫催化劑。采用BET、CO原位吸附、XRD、XPS等手段及催化劑加氫實驗表征和評價所制備的催化劑,并與器內預硫化法制備的催化劑對比,為制備免焙燒、分步預硫化催化劑提供參考。
1 實驗部分
1.1 試劑
MoO3,純度大于99.50%;堿式碳酸鎳,Ni質量分數高于44.00%;磷酸,w(H3PO4)≥85%;檸檬酸,純度大于99%;硫脲,分析純;聚乙二醇-200(PEG-200),分析純,沈陽市新化試劑廠產品。3%預硫化液(CS2體積分數為3%的航空煤油),自制。催化裂化柴油,中國石化鎮海煉化分公司提供。
1.2 催化劑的制備
取MoO3160.4 g、堿式碳酸鎳45.9 g、磷酸59.3 g及去離子水200 mL放入燒瓶中,置于山東鄄城華魯電熱儀器有限公司SHT數量調溫攪拌電熱套中,在設置溫度170℃、溶液實際溫度100℃下反應3 h。過濾,向濾液中加入70%(占Mo、Ni金屬氧化物質量和)檸檬酸,充分攪拌使其溶解。將溶液平均分成兩份,一份直接定容250 mL,得到Mo/Ni質量比為6的金屬浸漬液A;另一份加入40%(占Mo、Ni金屬氧化物質量和)PEG-200后,定容250 mL得到溶液B。
取溶液A加入5%(占Mo、Ni金屬氧化物質量和)的硫脲,再取溶液B三份分別加入0、5%和20%的硫脲。將上述4種溶液分別飽和浸漬60 mL載體,浸漬后載體編號為1、2、3、4號。將2號載體110℃烘干3 h,程序升溫至200℃焙燒2 h,400℃焙燒4 h制得催化劑MNAP401;1、3、4號3個樣品置于沈陽施博達儀器儀表有限公司微型水熱反應釜中,在150℃下進行原位沉淀反應6 h,取出后在110℃下烘干3 h,分別制得器外預硫化催化劑MNAL-5、MNAP40-5和MNAP40-20。
取催化劑MNAP401和MNAP40-20,以3%CS2(質量分數)的航空煤油為硫化劑,在反應壓力6 MPa、氫/油體積比600、空速3.2 h-1條件下,程序升溫在90、170、240℃分別停留3 h,370℃停留10 h,進行器內預硫化,制得器內預硫化催化劑MNAP40和分步預硫化催化劑MNAP40-202。
1.3 器外預硫化原位沉淀反應機理
當硫脲作為沉淀劑,在水熱反應中,硫脲中的S充當了反應的還原劑,原料中的S被氧化,生成SO42-;同時,它又是優良的硫化劑,為反應提供了硫源,生成MoS2。實際上,硫脲[CS(NH2)2]和水反應可分解為CO2、H2S和NH3[20],如式(1)所示,生成的H2S還原性很強,可以將MoO42-中的Mo6+還原為Mo4+,可能的反應如式(2)所示。
2NH3(g)+H2S(g)+CO2(g)
(1)
4MoS2+SO42-+6SCN-+24NH3(g)+9CO2(g)
(2)
在催化劑XRD表征中檢測出N2H6SO3的特征衍射峰,推測為反應中生成的副產物,可能的反應如式(3)所示。
N2H6SO3+2SCN-+3H2O
(3)
此外,硫脲分解生成的NH3和CO2少部分溶于水得 (NH4)2CO3,大部分以氣體形式離開了反應體系。
1.4 催化劑的活性評價
采用沈陽施博達儀器儀表有限公司連續進料的固定床微型反應器評價催化劑的加氫催化性能。催化劑裝填量20 mL,在壓力6 MPa、溫度370℃、氫/油體積比600、液體質量空速1.6 h-1的條件下,對劣質催化裂化柴油進行加氫。穩定10 h后開始取樣,之后每隔4 h取一次樣,直到40 h結束。原料催化裂化柴油的性質見表1。

表1 加氫實驗用催化裂化柴油的性質Table 1 Properties of the FCC diesel fraction for hydrotreating
1.5 催化劑的表征方法
采用美國Micromeritics公司ASAP 2420型物理吸附儀測量催化劑孔性質,樣品先在90℃下脫氣30 min,再200℃下脫氣2 h,以N2作為吸附質,在液氮(-196℃)下進行測定。采用日本理學公司 D/2500 型X-射線衍射儀測定樣品晶相結構(XRD),光源CuKα,波長0.154 nm,管工作電壓40 kV,工作電流80 mA。采用Thermo公司Nicolet 6700型傅里葉變換紅外光譜儀測定催化劑的CO原位吸附紅外光譜,常壓,500℃,高純氫氣還原3 h,300℃高真空凈化,室溫吸附CO,向原位池中引入少量CO氣體,吸附平衡30 min后,脫附至0.1 mPa。采用美國Perkin-Elmer Physics Electronics公司PIH 5300 X-射線光電子能譜儀獲取催化劑的XPS譜,MgKaX射線單色化激發源,功率150 W,荷電效應用來自載體Al2O3的Al(2p)峰(74.7 eV)校正。采用日本電子光學公司JEM-2000 型高分辨透射電鏡進行TEM分析,實際放大倍數30萬倍,加速電壓200 kV,LaB6燈絲,線分辨率0.14,點分辨率0.23 nm。催化劑樣品均保存于乙醇溶液中以避免與空氣接觸,每次測量時取少量催化劑于瑪瑙研缽中研細后超聲波分散于乙醇溶液中,取少量的懸浮液置于涂炭銅篩網上制樣,進行分析。
2 結果與討論
2.1 PEG-200及硫脲用量對器外預硫化催化劑性質的影響
考察了PEG-200及硫脲的加入量對器外預硫化催化劑MNAL-5、MNAP40-5和MNAP40-20的孔性質和金屬負載量的影響,結果列于表2。
從表2可見,與MNAL-5相比較,MNAP40-5的最可幾孔徑有所減小,但比表面積和孔容分別增大了57 m2/g和0.11 m3/g,且活性金屬負載量增加,說明PEG-200有擴孔及改善金屬分散性的作用;MNAP40-20孔容增加了0.07 cm3/g,比表面積增加了43 m2/g,說明硫脲的加入,一方面可以提供預硫化的硫來源,另一方面,可以對催化劑起到擴孔的作用。
圖1為器外預硫化催化劑MNAL-5、MNAP40-5和MNAP40-20的最可幾孔徑。由圖1可以看出,3種催化劑均出現了雙介孔結構,既存在提供加氫反應活性位的小孔,也存在為大分子反應物和產物提供擴散作用的大孔,這種等級介孔的存在有助于提高催化劑的加氫反應催化活性。
圖2為器外預硫化催化劑MNAL-5、MNAP40-5和MNAP40-20的N2吸附-脫附等溫線。由圖2可以看出,3種催化劑的N2吸附-脫附等溫線均屬于類型Ⅳ[21],都具有在較低相對壓力(p/p0)區曲線平穩向上、在較高p/p0區等溫線迅速上升的特點。從滯后環的形狀來看, MNAL-5 的大部分吸附存在于p/p0大于0.8的高壓區,說明此催化劑的孔道均以較大介孔為主,但吸附量較其他兩個催化劑少;MNAP40-20和MNAP40-5滯后環在p/p0為0.45處就開始迅速上升,一直持續到p/p0為1.0時,說明其孔道均屬于介孔范圍,且活性金屬在催化劑表面以多層吸附狀態存在。
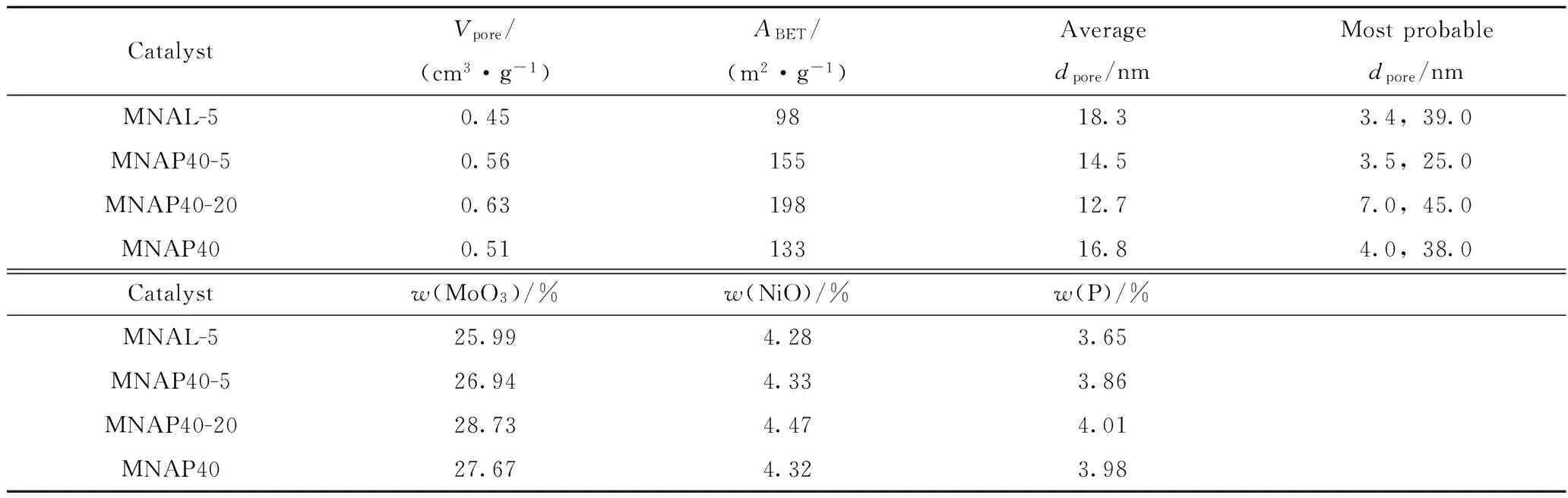
表2 器外預硫化催化劑MNAL-5、MNAP40-5和MNAP40-20的孔結構性質和金屬負載量Table 2 The texture properties and metal loadings of MNAL-5,MNAP40-5 and MNAP40-20 catalysts
Vpore—Pore volume;ABET—Specific surface area;dpore—Pore diameter.
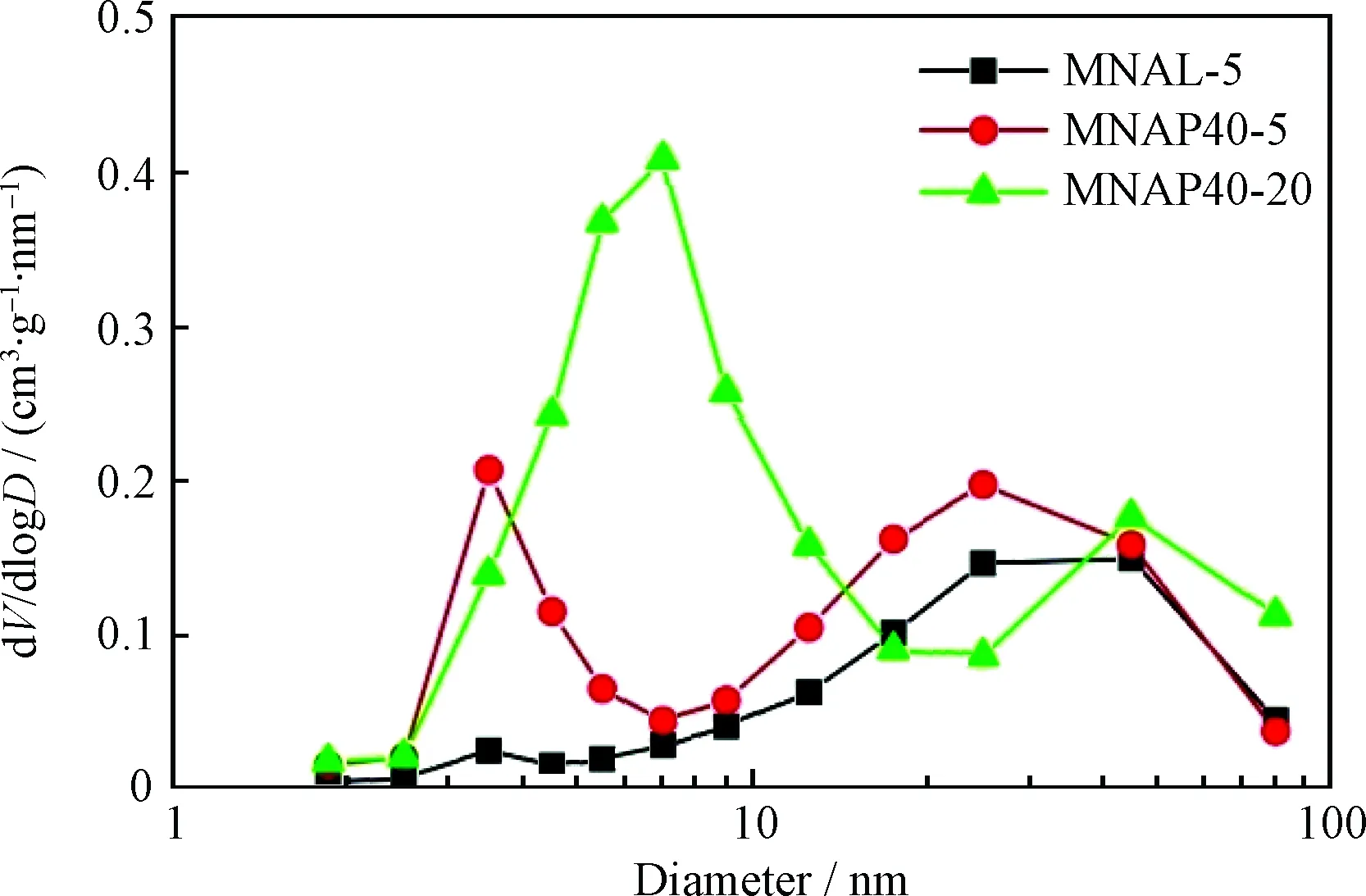
圖1 器外預硫化催化劑MNAL-5、MNAP40-5和 MNAP40-20的最可幾孔徑Fig.1 Most probable pore of MNAL-5, MNAP40-5 and MNAP40-20 catalysts
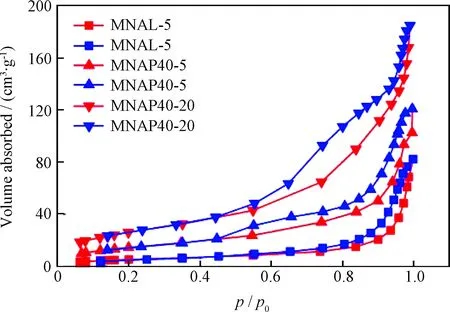
圖2 器外預硫化催化劑MNAL-5、MNAP40-5和 MNAP40-20的N2吸附-脫附等溫線Fig.2 N2 adsorption-desorption isothermals of MNAL-5, MNAP40-5 and MNAP40-20 catalystsAdsorption curve; Desorption curve
2.2 所制備的Mo-Ni/γ-Al2O3催化劑的表征結果
1.創新管理機制。針對當前社會組織的管理現狀,必須堅持油田黨委和社會組織黨組織負責日常管理,業務主管部門、行業協會加強協調聯系的管理模式,改變過去沒有婆家或者“誰才是婆家”管理主體不明晰的局面,明確工作責任,落實工作職責,形成覆蓋全區、上下貫通的社會組織黨建工作管理體系。
2.2.1 XRD分析
圖3為MNAL-5、MNAP40-5和MNAP40-20催化劑的XRD譜。從圖3可見,在2θ為37.44°、45.78°和67.31°處(PDF#04-080)明顯出現了γ-Al2O3的(311)、(400)和(441)晶面的特征衍射峰,在2θ為14.53°、33.21°、39.5°和58.32°處(PDF#17-0744)出現了MoS2的(003)、(101)、(103)和(110)晶面的特征衍射峰,且隨著PEG-200的加入及硫脲含量的增加,它們的特征衍射峰越來越明顯,說明活性Mo4+的量越來越多;尤其是在2θ為14.53°和58.32°處MNAP40-20的MoS2特征衍射峰最強,且峰型彌散,說明此催化劑MoS2的含量高,并在載體的內、外表面分散均勻。
MNAL-5的XRD譜中,在2θ為36.52°和53.79°處(PDF#50-0739)出現了MoO2的(100)和(102)晶面的特征衍射峰,在2θ為23.01°和25.00°處(PDF#47-1081)出現了MoO3的(011)和(200)晶面的特征衍射峰,說明此催化劑中還存在氧化態Mo6+及Mo4+。此種現象是由于器外預硫化不充分導致。MNAP40-5和MNAP40-20樣品的這些衍射峰趨于消失,證明隨著PEG-200的加入和硫脲含量的增加,樣品中有更多的氧化態鉬轉變成硫化態鉬,催化加氫反應活性位增加;MNAP40-20在2θ為32.21°和48.84°處(PDF#12-0041)出現了NiS的(300)和(131)晶面的特征衍射峰,且隨著PEG-200的加入和硫脲含量的增加,這些特征衍射峰趨于彌散,說明具有更良好的金屬分散性。此外,MNAL-5在2θ為20.34°處(PDF#42-0659)出現了N2H6SO3的(112)晶面的特征衍射峰,但是MNAP40-5和MNAP40-20的此峰明顯減小甚至消失,與此同時Mo4+的特征衍射峰明顯增強,說明PEG-200的加入可以減少副產物的生成。
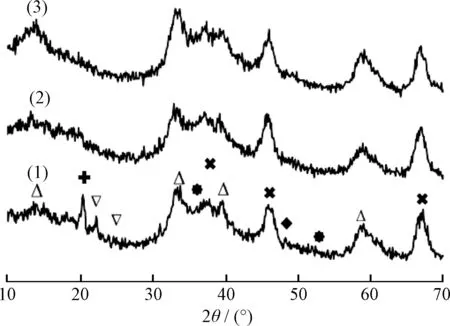
圖3 器外預硫化催化劑MNAL-5、MNAP40-5和 MNAP40-20催化劑的XRD譜Fig.3 XRD patterns of MNAL-5,MNAP40-5 and MNAP40-20 catalysts(1) MNAL-5; (2) MNAP40-5; (3) MNAP40-20 MoS2; MoS3; NiS; MoS2; γ-Al2O3; N2H6SO3
2.2.2 CO原位吸附紅外光譜
CO作為探針的紅外光譜被廣泛應用于催化劑表面活性金屬及其存在狀態的表征[22]。器外預硫化催化劑MNAL-5、MNAP40-5和MNAP40-20的CO原位吸收紅外光譜示于圖4。
圖4顯示,3種催化劑均產生了<2000 cm-1及>2000 cm-1的CO特征吸收峰,說明CO既有在Ni/Al2O3上的橋式吸附態[23],也有在Mo離子(Mo4+、Mo5+、Mo6+)上的線式吸附態。2187 cm-1處為CO在Mo4+上(Mo4+-CO)的吸附峰,2152 cm-1為CO在Mo5+上(Mo5+-CO)的吸附峰,2084 cm-1處接近于CO在純Ni硫化相(Ni2+-CO)上的吸附峰[24],2036 cm-1為CO在Mo6+上(Mo6+-CO)的吸附峰,1967 cm-1及1922 cm-1為CO在Ni/Al2O3上的橋式吸附的吸附峰。隨著PEG-200的加入及硫脲含量的增加,在2187 cm-1處CO吸附于Mo4+的峰和在2084 cm-1處CO吸附于Ni硫化相的峰均逐漸增強,而在2036 cm-1處CO吸附于Mo6+的峰和在2152 cm-1處CO吸附于Mo5+的峰均逐漸減弱,說明催化劑中有更多的Mo6+轉變成活性Mo4+,也有更多的氧化態Ni轉變成硫化態Ni,使催化劑活性位增加。

圖4 MNAL-5、MNAP40-5和MNAP40-20的 CO原位吸附紅外光譜Fig.4 FT-IR spectra of CO adsorbed for MNAL-5, MNAP40-5 and MNAP40-20 catalysts(a) MNAL-5; (b) MNAP40-5; (c) MNAP40-20
2.2.3 XPS表征
XPS分析能得出活性組分的種類、存在狀態、在催化劑表面的分散狀態及其與載體的作用方式等信息[25]。圖5為硫化態催化劑MNAP40-20、MNAP40和MNAP40-202的Mo3d的XPS譜。

圖5 硫化態催化劑MNAP40-20、MNAP40和 MNAP40-202的Mo3d XPS譜Fig.5 Mo3d XPS profiles of MNAP40-20, MNAP40 and MNAP40-202 catalysts(a) MNAP40-20; (b) MNAP40; (c) MNAP40-202
由圖5可知,它們均存在2種價態的鉬物種。主峰的結合能分別在232.0、231.7和232.0 eV,可歸屬于 +6 價的MoO3(Mo3d3/2結合能為232.5 eV)、Al2(MoO4)3(Mo3d3/2結合能為231.7 eV)或NiMoO4(Mo3d3/2結合能為232.8 eV);另一峰的結合能分別為229.0、228.5和228.8 eV可歸屬于+4價的MoS2(Mo3d5/2結合能為228. 9 eV)或低價鉬的氧硫物種。所得硫化后主峰的結合能數值比理論值稍小,是因為硫化作用可使鉬物種趨向于生成MoO2或者正六價鉬的硫化形式[26]。
圖6為硫化態催化劑MNAP40-20、MNAP40和MNAP40-202的Ni2p的XPS譜。結合能856.2 eV處的峰歸屬于與氧化鋁相互作用的Ni2+物種,854.0 eV處歸屬于NiMoS相Ni(NiMoS)的結合能,853.2 eV處歸屬于Ni的硫化物。

圖6 硫化態催化劑MNAP40-20、MNAP40和 MNAP40-202的Ni2p XPS譜Fig.6 Ni2p XPS profiles of MNAP40-20, MNAP40和MNAP40-202 catalysts(a) MNAP40-20; (b) MNAP40; (c) MNAP40-202
表3為XPS測得的MNAP40-20、MNAP40和MNAP40-202不同價態的Mo、Ni表面原子比和相對含量。從表3可見, MNAP40-20的Mo4+(MoS2)的峰面積占鉬物種總體比例的74.8%,證明催化劑在制備過程中已完成第一步器外預硫化的過程,具有一定的活性硫化態Mo4+。MNAP40比MNAP40-20的硫化度高4%,說明在實驗條件下只經過器外預硫化的硫化效果沒有器內硫化的硫化效果好。但是,經過分步預硫化后,MNAP40-202催化劑的Mo4+相對含量最高,達81.4%,比直接器內預硫化催化劑MNAP40的硫化度高2.6百分點,說明采用的分步預硫化法制備的催化劑具有較高的硫化度。此外,MANP40-202有最高的NiMoS比例,分別比MNAP40及MNAP40-20
高5.7百分點及6.6百分點。邊緣鑲嵌Ni顆粒的MoS2形成的NiMoS相的邊角具有加氫脫硫(HDS)和加氫脫氮(HDN)活性[27],說明MNAP40-202應具有最高的脫硫及脫氮催化活性。由表3還可知, MNAP40-202的Mo/Al及Ni/Al比最低。說明其表面與載體氧化鋁作用的Mo及Ni的比例最低,而獨立Mo、Ni硫化物的數量將隨之增加[27],此催化劑應該具有最高的催化活性,這與加氫評價結果相吻合。
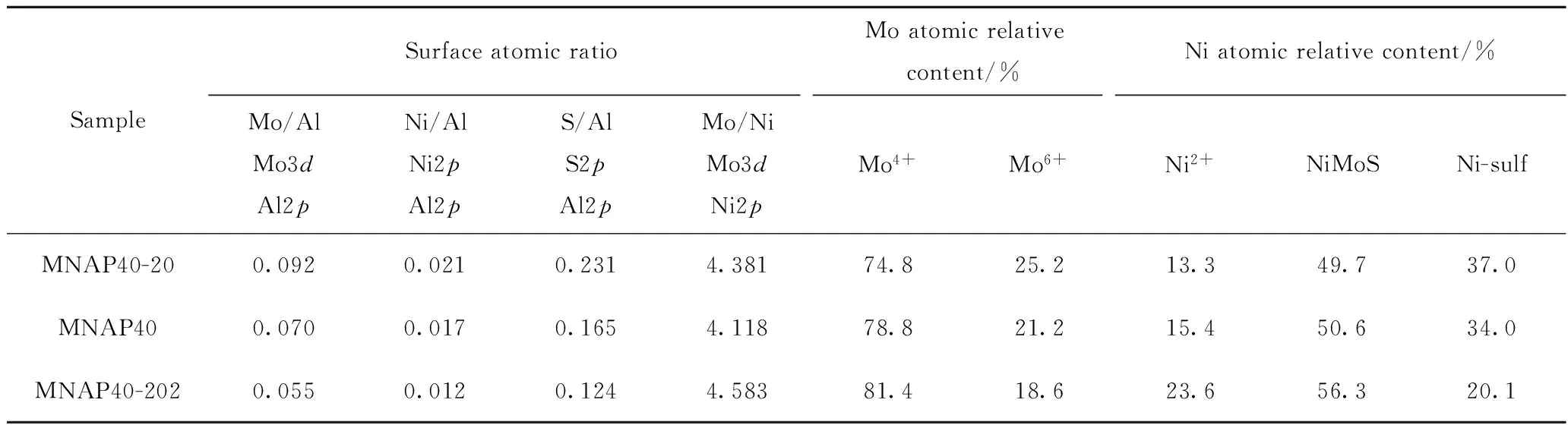
表3 MNAP40-20、MNAP40和MNAP40-202中不同價態的Mo、Ni表面原子比和相對含量Table 3 Surface atomic ratio and content of different valences Mo, Ni specie for MNAP40-20, MNAP40 and MNAP40-202 catalysts
2.2.4 TEM分析
圖7為硫化態催化劑MNAP40-20、MNAP40和MNAP40-202的TEM照片。圖7中,平行黑線間距為0.61 nm左右,是典型的MoS2晶面間距[28-29],多層黑線代表多層MoS2結構。3個樣品均有平行黑線層,可確認活性金屬在各催化劑表面均有效負載,且均為多層堆積,此現象與前面的BET表征相吻合。其中, MNAP40-20和MNAP40表面活性金屬平均堆積層數分別為2~3層和3~6層,平均長度分別8~10 nm和6~8 nm,而MNAP40-202表面活性金屬的堆積層數很高,甚至達到6~8層,而平均長度達8~10 nm。MoS2堆積層數的增加能夠消除平躺吸附的反應物分子的空間位阻,有利于加氫反應的進行[27]。一般HDS反應發生在MoS2的角位,HDN反應發生在MoS2的邊位和角位,在一定程度內高的金屬堆積層數有利于HDS和HDN反應的發生,相比之下MNAP40-202應具有最高的加氫脫硫、脫氮催化活性。

圖7 MNAP40-20、MNAP40和MNAP40-202的TEM照片Fig.7 TEM images of MNAP40-20, MNAP40 and MNAP40-202 catalysts(a) MNAP40-20; (b) MANP40; (c) MANP40-202
2.3 MNAP40-20、MNAP40和MNAP40-202的加氫反應催化活性
MNAP40-20、MNAP40及MNAP40-202加氫反應催化活性示于圖8。

圖8 MNAP40-20、MNAP40和MNAP40-202 催化加氫活性隨時間的變化Fig.8 Hydrogenation performances of MNAP40-20, MNAP40 and MNAP40-202 catalysts vs reaction time(a) HDS performance; (b) HDN performance;(c) HDA performance Reaction conditions:Temperature of 370℃; Liquid hourly space velocity of 1.6 h-1; Pressure of 6 MPa
由圖8(a)可看出,經3種預硫化方法處理的柴油加氫催化劑的脫硫率最終都趨于穩定,其平均脫硫活性從高到底的順序為MNAP40-202、MNAP40、MNAP40-20,且分步硫化法制備的催化劑MNAP40-202在40 h反應結束時脫硫率穩定在97%;而只器外預硫化的催化劑MNAP40-20的脫硫率最終穩定在93.66%,其脫硫率在4~16 h出現了下降的趨勢后又逐漸上升,可能由于預硫化不完全,反應中活性金屬被生成的H2S對催化劑起到了一定的硫化作用造成的;器內預硫化的催化劑MNAP40的最終脫硫率穩定在94.72%。上述結果證明,分步預硫化的催化劑在柴油加氫脫硫反應中的活性高于直接器內預硫化法制備的催化劑。由圖8(b)可以看出,MNAP40-202、MNAP40及MNAP40-20 3種催化劑的40 h柴油加氫脫氮率變化趨勢基本相同,最終脫氮率分別穩定在89.32%、87.38%及75.93%,說明分步預硫化的催化劑加氫脫氮活性也高于直接器內預硫化處理的催化劑。如圖8(c)所示,分步預硫化的催化劑MNAP40-202和直接器內預硫化的催化劑MNAP40均具有良好的芳烴飽和能力,在反應16 h后趨于穩定。MNAP40-202最終穩定的轉化率達到42.75%,稍高于催化劑MNAP40的39.29%;而只器外預硫化的催化劑MNAP40-20的芳烴轉化率隨著時間的延長,其活性逐漸減弱,40 h后轉化率為37.65%。此現象可能由于器外預硫化法的預硫化液黏度過大,在本實驗條件下預硫化不完全,催化劑的初始反應活性低,大分子芳烴生成的較大有機產物覆蓋催化劑表面造成孔道部分堵塞[30-31],降低了最終催化劑處理芳烴大分子的能力。
3 結 論
(1)利用原位沉淀法制備分步預硫化催化劑過程中, PEG-200以及硫脲的加入可以改善此系列催化劑的孔結構。當硫脲加入量為20%時,MNAP40-20的孔性質最好,其孔容為0.63 cm3/g、最可幾孔徑為7.0和45.0 nm、比表面積為198 cm2/g,并且具有較好的金屬分散性。
(2)原位沉淀法制備分步預硫化催化劑在制備過程中已有74.8%的Mo被硫化,此時催化劑已經具有催化活性,但比器內預硫化催化劑的硫化度低,而經分步預硫化后催化劑硫化度高達81.4%。
(3)催化加氫反應40 h后穩定,原位沉淀法制備的分步預硫化催化劑催化加氫所得脫硫率、脫氮率和芳烴飽和率分別達到97.00%、89.32%和42.75%,比直接器內預硫化催化劑催化所得的分別增加了2.28百分點、1.94百分點和3.46百分點,說明此方法處理的催化劑具有較高的加氫催化性能及穩定性,為制備免焙燒、分步預硫化催化劑的方法提供一定的科學依據。
[1] LIU Y D, GAO L, WEN L Y. Recent advance in heavy oil hydroprocessing technologies[J].Recent Patents on Chemical Engineering, 2009, 2(1): 22-36.
[2] MOHAN S R, JORGE A, DIAZ J A. A review of recent advances on process technologies for upgrading of heavy oils and residua[J].Fuel, 2007, 86(9): 1216-1231.
[3] SUNHWAN H, JOONGWON L, JEONG G S. Pd catalyst supported on SiO2-A12O3xero gel for hydrocracking of paraffin wax to middle distillate[J].Journal of Industrial & Engineering Chemistry, 2011, 17(2): 310-315.
[4] JOONGWON L, SUNHWAN H, SANG B L, et al. Production of middle distillate through hydrocracking of paraffin wax over NiMo/TiO2-SiO2catalysts[J].The Korean Journal of Chemical Engineering, 2010, 27(6): 1755-1759.
[5] 張志民, 郭長友, 凌鳳香, 等. 氧化鋁表面鈦改性的機理分析[J].石油煉制與化工, 2012, 43(10): 49-54. (ZHANG Zhimin, GUO Changyou, LING Fengxiang, et al. Mechanism analysis of Ti-modifide alumina support[J].Petroleum Processing and Petrochemicals, 2012, 43(10): 49-54.)
[6] SUNHWAN H, JOONGWON L, SUNYANG P. Production of middle distillate through hydrocracking of paraffin wax over NiMo/SiO2-A12O3catalysts: Effect of SiO2-A12O3composition on acid property and catalytic performance of NiMo/SiO2-AI2O3catalysts[J].Catalysis Letters, 2009, 129(1/2): 163-169.
[7] 王鼎聰. 納米自組裝合成大孔容介孔氧化鋁[J] .中國科學, 2009, 39(5): 420-431. (WANG Dingcong. Mesoporous aluminum oxide support with large pore volume by nano self-assembly[J].Science in China, 2009, 39(5): 420-431.)
[8] 田野, 趙德智, 王鼎聰, 等. 自組裝催化劑在混合油加氫精制中的應用[J].石油化工高等學校學報, 2014, 27(2):15-20. (TIAN Ye, ZHAO Dezhi, WANG Dingcong, et al. The application of self-assembly catalyst in hydrorefining performance for mixed oil[J].Journal of Petrochemical Universities, 2014, 27(2): 15-20.)
[9] 鄢景森, 王海彥, 張靜茹, 等. TiO2-Al2O3載體的制備方法對其負載的磷化鎳催化劑加氫脫氮反應性能的影響[J].物理化學學報, 2014, 30(7): 1309-1317. (YAN Jingsen, WANG Haiyan, ZHANG Jingru, et al. Effect of TiO2-Al2O3support preparation technique on hydrodenitrogenation of Ni2P/TiO2-Al2O3catalysts[J].Acta Physico-Chimica Sinica, 2014, 30(7): 1309-1317.)
[10] 柴永明, 安高軍, 柳云騏, 等. 過渡金屬硫化物催化劑催化加氫作用機理[J].化學進展, 2007, 19(2/3): 234-242. (CHAI Yongming, AN Gaojun, LIU Yunqi, et al. Transition metal sulfides hydrogenation catalysts: Active phase structure and mechanism of the catalytic reaction[J].Progress in Chemistry, 2007, 19(2/3): 234-242.)
[11] 高玉蘭, 方向晨. 加氫處理催化劑器外預硫化技術研究與展望[J].化工進展, 2010, 29(3): 465-471. (GAO Yulan, FANG Xiangchen. Progress and prospect in ex-situ presulfurization for hydrotreating catalyst[J].Chemical Industry and Engineering Progress, 2010, 29(3): 465-471.)
[12] 羅樹權, 孫征, 高雪. 加氫催化劑器外預硫化技術現狀[J] . 化工技術與開發, 2014, 8(3): 34-37. (LUO Shuquan, SUN Zheng, GAO Xue. Current situation of ex-situ pre-sulfiding hydrotreatment catalyst[J].Technology & Development of Chemical Industry, 2014, 8(3): 34-47.)
[13] 丁慶玉, 于春梅, 王燕. 加氫催化劑器外預硫化技術研究[J].化學工程師, 2013, 27(7): 70-73. (DING Qingyu, YU Chunmei, WANG Yan. Research of off-site presulfurization hydrotreating catalysts[J].Chemical Engineer, 2013, 27(7): 70-73.)
[14] 李童, 董群, 馮熙桐. 加氫催化劑預硫化技術研究進展[J].化學工程師, 2014, 28(1): 42-44. (LI Tong, DONG Qun, FENG Xitong. Research progress in the hydrogenation catalyst presulfrization[J].Chemical Engineer, 2014, 28(1): 42-44.)
[15] GAO Y, FANG X, CHENG Z. A comparative study on the ex situ and in situ presulfurization of hydrotreating catalysts[J].Catalysis Today, 2010, 158: 496-503.
[16] 夏遠亮. 原位分解法制備免預硫化CoMoS/γ-Al2O3催化劑的表征及加氫性能研究[J].分子催化, 2008, 22(3): 224-229. (XIA Yuanliang. Study on characterization and hydrogenization performance of CoMoS/γ-Al2O3, catalyst prepared by in-situ decomposition method[J].Journal of Molecular Catalysis (China), 2008, 22(3): 224-229.)
[17] 林凌, 伊曉東, 邱波, 等. 免預硫化加氫脫硫MoNiP/Al2O3催化劑的制備和表征[J].催化學報, 2007, 28(12): 1096-1100. (LIN Ling, YI Xiao-dong, QIU Bo, et al. Preparation and characterization of catalyst for tiophene presulfidation-free MoNiP/Al2O3hydrodesulfurization[J].Chinese Journal of Catalysis, 2007, 28(12): 1096-1100.)
[18] 田維乾, 劉靜, 劉燦, 等. CoMoS/γ-Al2O3催化劑對麻瘋樹油加氫處理的研究[J].燃料化學學報, 2013, 41(2): 207-213. (TIAN Weiqian, LIU Jing, LIU Can, et al. Hydrotreatment of jatropha oil over CoMoS/γ-Al2O3catalyst[J].Journal of Fuel Chemistry and Technology, 2013, 41(2): 207-213.)
[19] 任春曉, 吳培, 李振昊, 等. 加氫催化劑預硫化技術現狀[J].化工進展, 2013, 32(5): 1060-1064. (REN Chunxiao, WU Pei, LI Zhenhao, et al. The status of presulfurization technology for hydrogenation catalyst[J].Chemical Industry and Engineering Progress, 2013, 32(5): 1060-1064.)
[20] YU S H, WU Y S, YANG J, et al. A novel solventothermal synthetic route to nanocrystalline CdE (E=S, Se, Te) and morphological control[J].Chemistry of Materials, 1998, 10(10): 2309-2312.
[21] 吳雨航, 凌鳳香, 趙國利, 等. CoMo/Al2O3-SiO2催化劑原位紅外光譜研究[J].當代化工, 2015, 44(5): 962-965. (WU Yuhang, LING Fengxiang, ZHAO Guoli, et al. In-situ FT-IR Study on the CoMo/Al2O3catalyst[J].Contemporary Chemical Industry, 2015, 44(5): 962-965.)
[22] DELIY I V, VLASOVA E N, NUZHDIN A L, et al. Hydrodeoxygenation of methyl palmitate over sulfided Mo/Al2O3, CoMo/Al2O3and NiMo/Al2O3catalysts[J].Rsc Advances, 2013, 4(5): 2242-2250.
[23] 張玉涵, 凌鳳香, 王少軍, 等. Co-Mo/γ-Al2O3催化劑的原位紅外光譜表征研究[J].燃料化學學報, 2013, 41(6): 710-714.(ZHANG Yuhan, LING Fengxiang, WANG Shaojun, et al. An in-situ FT-IR study on the CO and NO co-adsorption on the Co-Mo/γ-Al2O3catalysts[J].Journal of Fuel Chemistry and Technology, 2013, 41(6): 710-714.)
[24] HADJIVANOV K I, VAYSSILOV G N. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule[J].Cheminform, 2003, 34(17): 307-511.
[25] 邱麗美, 齊和日瑪, 劉清河, 等. X射線光電子能譜法研究加氫脫硫催化劑中活性元素的化學態[J].石油學報(石油加工), 2011, 27(4): 638-642. (QIU Limei, Qiherima, LIU Qinghe, et al. Investigation of chemical states for hydrodesulfurization catalysts by using X-Ray photoelectron spectroscopy[J].Acta Petrolei Sinica(Petroleum Processing Section), 2011, 27(4): 638-642.)
[26] 朱崇業, 牛國興, 陳海鷹, 等. 鉬鎳系列加氫處理催化劑的表面活性結構[J].復旦學報(自然科學版), 1995, 34(5): 490-500.(ZHU Chongye, NIU Guoxing, CHEN Haiying, et al. Surface species structure of nickel-molybdenum hydrotreating catalyst[J].Journal of Fudan University (Natural Science), 1995, 34(5): 490-500.)
[27] 周同娜, 尹海亮, 柳云騏,等. 磷含量對NiMo/γ-Al2O3催化劑活性相結構的影響[J].燃料化學學報, 2010, 38(1): 69-74. (ZHOU Tongna, YIN Hailiang, LIU Yunji, et al. Effect of phosphorus on the interaction between active component and carrier of NiMo/Al2O3catalysts[J].Petroleum Processing and Petrochemicals, 2010, 38(1): 69-74.)
[28] FERDOUS D, DALAI A K, ADJAYE J, et al. Surface morphology of NiMo/Al2O3catalysts incorporated with boron and phosphorus: Experimental and simulation[J].Applied Catalysis A General, 2005, 294(1): 80-91.
[29] HUIRACHE-ACUA R, ALBITER M A, ESPINO J, et al. Synthesis of Ni-Mo-W sulphide catalysts by ex situ decomposition of trimetallic precursors[J].Applied Catalysis A General, 2006, 304(1): 124-130.
[30] FURIMSKY E, MASSOTH F E. Deactivation of hydroprocessing catalysts[J].Catalysis Today, 1999, 52(52): 381-495.
[31] LEBRETON R, BRUNET S, PéROT G, et al. Deactivation and characterization of hydrotreating NiMo/Al2O3catalyst coked by anthracene[J].Studies in Surface Science & Catalysis, 1999, 126(99): 195-201.
Preparation, Characterization and Catalytic Hydrotreating Performance ofStepped Presulfurized Mo-Ni/γ-Al2O3Catalyst
ZHANG Qiang1, DING Wei1,3, WANG Dingcong2, ZHAO Dezhi1, YANG Chensi1, ZHANG Zhiwei1
(1.CollegeofChemicalEngineeringandEnvironmentalEngineering,LiaoningUniversityofPetroleum&ChemicalTechnology,Fushun113001,China; 2.FushunResearchInstituteofPetroleumandPetrochemicals,SINOPEC,Fushun113001,China;3.StateKeyLaboratoryofHeavyOilProcessing,ChinaUniversityofPetroleum,Beijing102249,China)
The prevulcanization precipitation liquid was prepared by self-assembly with thiocarbamide, citric acid and polyethylene glycol-200. The two-stepped presulfurization Mo-Ni/Al2O3catalysts were prepared by in-situ precipitation method and characterized by BET, XRD, FT-IR of CO adsorbed, XPS, and TEM. The hydrogenation performances of the prepared catalyst were evaluated with micro fixed bed reactor and with the directly in-situ presulfurization catalyst as reference. The results showed that the two-stepped presulfurization Mo-Ni/Al2O3already achieved 74.8% of sulfidity after the first step of presulfurization (ex-situ presulfurization). The additions of polyethylene glycol-200 and thiocarbamide made an improvement of the pore properties of the catalysts. The biggest specific surface of 198 cm2/g was obtained when the mass fraction of thiocarbamide reached 20% of Mo and Ni oxides in the catalyst, at the same time, the active metal dispersion was better and the sulfidity was 2.60 percentage points higher than the reference catalyst. The hydrodesulfurization (HDS), hydrodenitrogenation (HDN) and hydrodearomatization (HDA) rates of 40 h hydrotreating for FCC diesel fraction over the two-stepped presulfurization Mo-Ni/γ-Al2O3catalyst were 2.28 percentage points, 1.94 percentage points and 3.46 percentage points higher than those over the reference catalyst, respectively.
two-stepped presulfurization; in-situ precipitation; catalysts; hydrodesulfurization (HDS); hydrodenitrogenation (HDN)
2016-01-15
中國石油化工集團公司資助項目(總合-JQ1416)和中國海洋石油總公司資助項目(20140331)基金資助
張強,男,碩士研究生,主要從事加氫工藝及其催化劑研究
丁巍,女,講師,博士研究生,從事重質油加氫催化劑研發;E-mail:cicy1125@163.com
1001-8719(2017)01-0032-10
TQ426.95
A
10.3969/j.issn.1001-8719.2017.01.005
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
