SILAR法制備TiO2/CdS/Co-Pi水氧化光陽極及其性能
2016-12-29 05:42:51劉歡歡楊玉林強亮生
物理化學(xué)學(xué)報 2016年11期
周 麗 劉歡歡 楊玉林 強亮生
(哈爾濱工業(yè)大學(xué)化工與化學(xué)學(xué)院,哈爾濱150001)
SILAR法制備TiO2/CdS/Co-Pi水氧化光陽極及其性能
周 麗*劉歡歡 楊玉林 強亮生
(哈爾濱工業(yè)大學(xué)化工與化學(xué)學(xué)院,哈爾濱150001)
采用連續(xù)離子層吸附法(SILAR)沉積CdS制備type-II異質(zhì)結(jié)TiO2/CdS光陽極,用光電化學(xué)沉積法在TiO2/CdS表面沉積催化劑(Co-Pi)得到TiO2/CdS/Co-Pi水氧化光陽極。通過X射線衍射(XRD)儀、掃描電子顯微鏡(SEM)、X射線光電子能譜(XPS)儀等對樣品結(jié)構(gòu)及組成進行分析,證明CdS與Co-Pi已成功負載在TiO2表面。用已制備的光陽極在中性溶液中模擬水氧化測試,在較低外偏壓(0 V(vs Ag/AgCl))和無電子犧牲劑的情況下,即使在可見光照射下,依然得到較高的初始光電流和穩(wěn)定光電流,分別為1.3和0.5 mA·cm-2,表明制備的光陽極可以在可見光照下有效地驅(qū)動水氧化反應(yīng)。光電化學(xué)池的工作原理為,CdS吸收光子產(chǎn)生光生電子-空穴,TiO2和Co-Pi分別傳輸電子和空穴,空穴進行水氧化,電子轉(zhuǎn)移到陰極完成質(zhì)子還原。
光催化;水氧化;連續(xù)離子層吸附及反應(yīng);可見光;TiO2/CdS/Co-Pi光陽極
1 引言
在自然界中,綠色植物能夠利用光合作用將水分解,進而將太陽能轉(zhuǎn)化為化學(xué)能1,2,其光引發(fā)的階段是產(chǎn)生長壽命的電荷分離態(tài)。受此啟發(fā),光敏半導(dǎo)體可以在光激發(fā)下產(chǎn)生電子和空穴的電荷分離態(tài),以光敏半導(dǎo)體構(gòu)建光電化學(xué)池(PEC)將水分解為H2和O23,4,理論上能夠可持續(xù)地將太陽能轉(zhuǎn)化為氫能。構(gòu)建PEC的半導(dǎo)體光電極材料需要滿足以下條件:具有良好的光穩(wěn)定性、在可見光區(qū)有較強的光譜響應(yīng)、導(dǎo)帶與價帶的能帶位置分別可以驅(qū)動水的氧化和質(zhì)子還原、光生載流子分離與傳輸速率能夠滿足電極表面化學(xué)反應(yīng)速率等5。基于上述條件,半導(dǎo)體的異質(zhì)結(jié)構(gòu)能將各種不同半導(dǎo)體的優(yōu)點集中到一個光電極上,使其展現(xiàn)出更優(yōu)異的性能。TiO2為n型半導(dǎo)體,可以有效地傳輸電子,并具有良好的化學(xué)穩(wěn)定性,同時CdS作為光捕獲材料,能將太陽光利用范圍拓展到可見光區(qū),兩者復(fù)合形成的type-II型異質(zhì)結(jié)界面能夠有效地提高光生空穴-電子的分離效率6,極大地提高了光陽極的光電化學(xué)性能7,例如,CdS敏化TiO2光陽極構(gòu)建的PEC能夠在電子犧牲劑存在的條件下將水中的質(zhì)子還原為氫氣8-12。在此基礎(chǔ)上,利用水氧化催化劑修飾半導(dǎo)體光陽極13-17,可以在進一步減少電子-空穴復(fù)合的同時以水作為電子給體,使太陽能制氫走向?qū)嵱没牡缆贰?008年,Nocera研究組18發(fā)表了基于金屬鈷的高效水氧化催化劑Co-Pi,以其來源廣泛、易制備、催化效果較強的特點迅速成為水分解領(lǐng)域的研究熱點19,20。盡管Co-Pi修飾光陽極的催化機理仍未被徹底解析,但目前普遍認可的解釋為,光敏半導(dǎo)體產(chǎn)生的光生空穴使鈷達高價氧化態(tài),進而能夠?qū)⑺肿友趸?/p>
目前,TiO2/CdS光陽極制備的方法較為繁瑣,并且在可見光驅(qū)動、沒有電子犧牲劑存在的條件下光電流響應(yīng)較弱21-23。Zhong等5利用水熱法制備TiO2納米線陣列,再利用化學(xué)氣相沉積在TiO2表面沉積CdS的方法制備TiO2/CdS/Co-Pi光陽極,該光陽極在0.6 V(vsAg/AgCl)的外偏壓下,用模擬自然光(含紫外光)驅(qū)動水分解,得到了0.8 mA·cm-2光電流響應(yīng)。光敏半導(dǎo)體材料的光電性能會由于其尺寸或形貌的不同而產(chǎn)生差異,因此為了考察其他制備方法對TiO2/CdS/Co-Pi光陽極的影響,本文采用市售的二氧化鈦(P25)制備光陽極坯片,通過SILAR法沉積CdS敏化TiO2,最后在其表面通過光輔助電沉積Co-Pi制備了TiO2/CdS/Co-Pi光陽極。該體系能夠在無電子犧牲劑的中性水相電解液中工作,在可見光驅(qū)動下將水分解。在較低的外偏壓下,本文所制備的TiO2/CdS/Co-Pi復(fù)合光陽極可以獲得較高且較穩(wěn)定的光電流。在TiO2/CdS/Co-Pi復(fù)合光陽極中,CdS能拓寬太陽的光利用范圍到達可見光區(qū),Co-Pi作為電極與電解液之間的空穴傳輸層,能夠加快電極的空穴向電解液中移動進而提高水氧化的效率,而TiO2納米顆粒能夠?qū)㈦娮觽鬏斨翐椒鶶nO2導(dǎo)電玻璃(FTO)進而到達對電極。
2 實驗部分
2.1試劑與儀器
硫化鈉、巰基丙酸、硝酸鎘、硝酸鈷、磷酸緩沖劑、乙基纖維素、松油醇、無水乙醇及無水甲醇均為市售分析純;導(dǎo)電玻璃(90%可見光透射率,15 Ω·m-2)購于日本NSG公司;納米二氧化鈦(P25)購于德國德固賽化學(xué)有限公司。
實驗光源為300 W氙燈附帶400 nm濾光片(CHF-XM-300W,北京暢拓科技有限公司);光電流測試和循環(huán)伏安采用電化學(xué)工作站(CHI 600E,上海辰華儀器公司);光陽極材料的形貌、組成分析采用掃描電子顯微鏡(SU1510,日本日立公司)、透射電子顯微鏡(FeiTecnia G2 STWIN,美國FEI公司)、X射線粉末衍射儀(Bruke D8Advance,德國布魯克公司)、X射線光電子能譜儀(AXIS Supra,日本島津企業(yè)有限公司);固體薄膜的紫外可見吸收光譜采用紫外可見近紅外分光光度計(PE Lambda S750,美國珀金埃爾默公司)。
2.2實驗過程
質(zhì)量分數(shù)(w)分別為16.8%(w)的TiO2、4.5% (w)的乙基纖維素以及78.7%(w)的松油醇混合攪拌48 h,配制成均勻的二氧化鈦漿料。通過絲網(wǎng)印刷的方法在2 cm×1 cm的導(dǎo)電玻璃表面印制1 cm× 1 cm面積的薄膜,100°C干燥5 min,重復(fù)印制四次。在空氣中程序升溫至500°C燒結(jié)后,自然冷卻,形成均勻的TiO2薄膜。
對已有文獻24中CdS的制備方法進行改進,將印有TiO2的FTO玻璃在0.1 mol·L-1巰基丙酸(MPA)的乙醇溶液中浸泡5 min后用乙醇洗滌,然后在0.1 mol·L-1Cd(NO3)2在乙醇溶液中浸泡5 min,之后用乙醇除去多余的Cd2+。吸附有Cd2+的TiO2薄膜在烘箱中干燥之后,最后在含有0.1 mol·L-1Na2S的甲醇和去離子水(體積比為3:7)的混合液中浸泡5 min,用甲醇除去多余的S2-,烘箱中烘干5 min。重復(fù)以上操作5次,得到吸附有CdS量子點的TiO2/CdS半導(dǎo)體薄膜。
稱取一定量的Co(NO3)2,使用pH值為6.85的磷酸緩沖溶液配制成0.5 mmol·L-1Co(NO3)2電解質(zhì)溶液,以Ag/AgCl電極作為參比電極,Pt為對電極,TiO2/CdS光陽極為工作電極,形成三電極體系,在可見光照射下,外加偏壓0.65 V,在工作電極表面沉積Co-Pi,形成TiO2/CdS/Co-Pi電極。
3 結(jié)果與討論
3.1復(fù)合光陽極的表征
研究SILAR法制備TiO2/CdS光陽極時,需要探索不同工藝條件對光陽極光電流響應(yīng)的影響,主要對SILAR法制備過程中的沉積液濃度、浸泡時間、TiO2層數(shù)以及SILAR循環(huán)次數(shù)這些工藝條件進行了優(yōu)化。在探索過程中,控制單一變量,每個循環(huán)中光照持續(xù)50 s,每個實驗重復(fù)兩個循環(huán),測試電流-時間曲線。結(jié)果如圖1所示,最佳的SILAR法制備工藝條件為0.1 mol·L-1的沉積液、浸泡時間為5 min、TiO2基底層數(shù)為4層、SILAR循環(huán)次數(shù)為5次。此時,TiO2/CdS光陽極的光電流響應(yīng)最大,最大瞬時電流為0.8 mA·cm-2,經(jīng)過兩個50 s的on+-off光照循環(huán)后,電流密度降為0.4 mA·cm-2,且趨于穩(wěn)定。
用掃描電子顯微鏡觀察處理不同階段的顆粒形貌,如圖2所示,以P25為原料,以絲網(wǎng)印刷法制備的TiO2光陽極表面為分散均勻的球形TiO2顆粒,平均粒徑在40 nm左右(圖2(a))。通過SILAR法將CdS沉積于TiO2薄膜之后(圖2(b)),表面形貌沒有發(fā)生明顯變化,但平均粒徑略微增大,到達50 nm左右,表明CdS以微小納米粒子的形式包覆在TiO2表面。通過光電化學(xué)沉積后的TiO2/CdS/Co-Pi光陽極(圖2(c)),粒徑和形貌均可觀察到變化,但無法觀測到明顯的硫化鎘和Co-Pi形貌。
由圖3(a)的TiO2/CdS/Co-Pi復(fù)合納米顆粒透射電鏡圖可知,由SILAR法制備的TiO2/CdS/Co-Pi復(fù)合納米顆粒為硫化鎘包覆二氧化鈦形成的核殼結(jié)構(gòu)納米顆粒,圖3(b)的高分辨透射電鏡顯示的間距為0.318和0.355 nm的晶格條紋分別對應(yīng)銳鈦礦相二氧化鈦的(101)晶面和六方晶系硫化鎘的(101)晶面,并且在透射電鏡中沒有發(fā)現(xiàn)明顯的Co-Pi顆粒,這是由于Co-Pi同時具有分子以及無機粒子的特性,其粒徑極小,且沒有固定的形貌25-27。
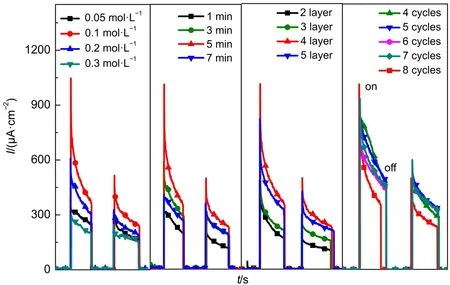
圖1 SILAR法的不同實驗條件對應(yīng)的電流密度-時間(I-t)電流圖Fig.1 I-t curves of different experimental processes with SILAR method

圖2 不同階段光陽極的掃描電鏡圖Fig.2 SEM images of photoanode at different stages

圖3 TiO2/CdS/Co-Pi復(fù)合納米顆粒的(a)透射電鏡圖和(b)高倍透射電鏡圖Fig.3 (a)Transmission electron microscopy(TEM)and(b)high resolution TEM of TiO2/CdS/Co-Pi composite nanoparticles
對制備的光陽極進行XRD測試表征,從圖4 (a)中可以看出對于TiO2/CdS/Co-Pi復(fù)合光陽極,在衍射角2θ為25.35°、47.98°及53.90°處出現(xiàn)了較強的衍射峰,這分別與銳鈦礦型TiO2的(101)、(200)、(105)晶面的特征衍射峰對應(yīng)(JCPDS No.21-1272)。在衍射角為26.50°、28.17°及47.79°處出現(xiàn)較弱的衍射峰,其對應(yīng)的分別為六方晶相CdS的(100)、(101)及(103)晶面特征衍射峰(JCPDS No.41-1049),導(dǎo)致CdS強度較弱的原因可能是SILAR法制備的CdS結(jié)晶度不高。

圖4 TiO2/CdS/Co-Pi光陽極的X射線衍射圖和X射線光電子能譜分析圖Fig.4 XRD and XPS survey scan from TiO2/CdS/Co-Pi photoanode
由于XRD無法檢測到Co-Pi的衍射峰,因此利用X射線光電子能譜(XPS)進一步分析TiO2/CdS/ Co-Pi光陽極上的組成和化學(xué)價態(tài),從圖4(b)的XPS全譜中可以看出樣品中存在Ti、O、Cd、S、P以及Co這幾種元素,沒有檢測到其他明顯的雜質(zhì)。圖4(c)為Co-Pi中P元素高分辨光電子能譜,其P 2p1/2電子結(jié)合能位置為133.48 eV,證實P以基團的形式存在。圖4(d)為Co元素的高分辨光電子能譜,電子結(jié)合能為781.21及797.43 eV的兩個峰分別對應(yīng)Co 2p3/2與Co 2p1/2出峰位置,表明Co主要以Co2+與Co3+的形式存在,并有一定的比例,不過在催化水氧化過程中兩者是存在相互轉(zhuǎn)化的,比例對催化的影響不大28。
用固體紫外-可見吸收光譜對TiO2/CdS/Co-Pi光陽極的光吸收性能進行研究,結(jié)果如圖5(a)所示,從圖中可看出單純的TiO2的吸收邊在400 nm左右,這與TiO2的能帶間隙(3.2 eV)一致8,而TiO2/ CdS光陽極的吸收邊在540 nm左右,由于CdS的能帶間隙為2.3 eV,以其作為光吸收層能將光陽極對太陽光的利用增加至可見光范圍,TiO2/CdS/Co-Pi與TiO2/CdS的吸收邊基本一致,這表明Co-Pi作為催化劑對光吸收影響不大,并且不會產(chǎn)生額外的帶隙躍遷。
由于TiO2/CdS/Co-Pi在可見光驅(qū)動下催化水氧化過程伴隨著電子轉(zhuǎn)移,因此,通過光電流響應(yīng)測試即可檢測復(fù)合光陽極的催化性能。以電化學(xué)工作站為平臺,TiO2/CdS/Co-Pi復(fù)合光陽極為工作電極,鉑電極為對電極,Ag/AgCl為參比電極,組裝光電化學(xué)池,在0 V(vs Ag/AgCl)偏置電壓下測試光電流,得出電流-時間曲線,如圖5(b)所示。沉積Co-Pi后相比于未沉積時的光電流瞬態(tài)電流增長至大約2倍,100 s后電流增長仍保持在25%左右。同時從圖中可以看出,經(jīng)過一段時間光照后TiO2/CdS/Co-Pi光陽極光電流密度有所衰減,這可能是由于CdS在光照條件下分解導(dǎo)致Co-Pi脫附造成的。但是,修飾后的光陽極性能得到顯著改善,證明TiO2/CdS/Co-Pi光陽極具有很好的水氧化效果。這個結(jié)果跟文獻5相比有了一定的提升,說明SILAR方法的有效性。
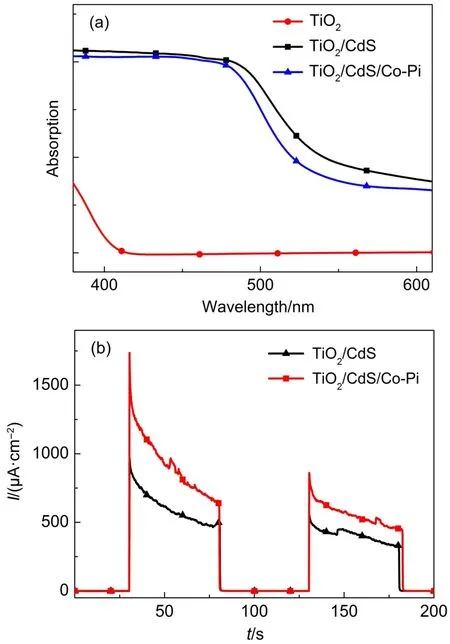
圖5 不同光陽極對應(yīng)的紫外-可見吸收曲線及沉積Co-Pi前后0 V(vsAg/AgCl)時對應(yīng)的電流-時間圖Fig.5 (a)UV-Vis absorption spectra of different photoanodes and(b)I-t curves of photoanode measured with and without deposition Co-Pi at 0 V(vsAg/AgCl)
3.2機理分析
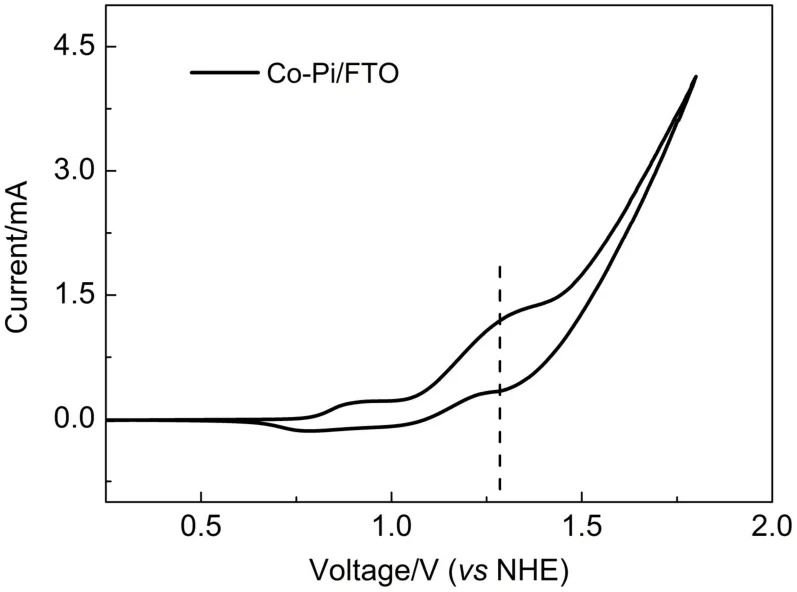
圖6 在FTO表面沉積Co-Pi對應(yīng)的循環(huán)伏安圖Fig.6 Cyclic voltammagram of FTO electrodeposition with Co-Pi
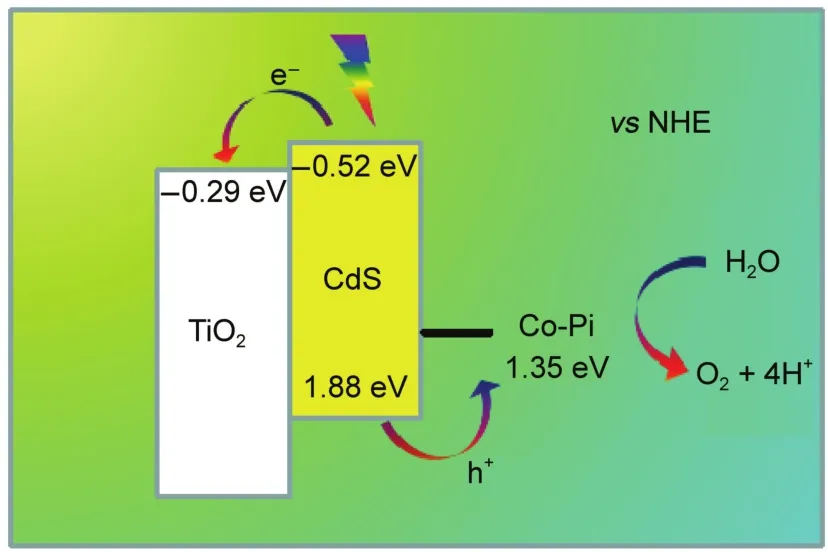
圖7 TiO2/CdS/Co-Pi光陽極進行水氧化反應(yīng)的實驗機理Fig.7 Mechanism schematic of TiO2/CdS/Co-Pi photoanode for water oxidation
首先將Co-Pi沉積在FTO玻璃表面,進行循環(huán)伏安測試(如圖6所示),Co-Pi在1.27 V(vs NHE)出現(xiàn)一個陽極氧化峰,隨后在電極表面產(chǎn)生一個極大的催化水氧化的現(xiàn)象。相對于中性條件下水的理論氧化電位0.82 V(vs NHE),催化水氧化的過電位為0.45 V,這極大地降低了實現(xiàn)水氧化所需的能量。根據(jù)TiO2、CdS與Co-Pi的導(dǎo)帶和價帶電位8建立了TiO2/CdS/Co-Pi光陽極工作機理圖(如圖7所示)。CdS在可見光激發(fā)下產(chǎn)生電子-空穴分離態(tài),光生電子從CdS的導(dǎo)帶傳遞到TiO2的導(dǎo)帶,而光生空穴從CdS的價帶傳遞到Co-Pi上,Co-Pi作為電極與電解液之間的空穴傳輸層,能夠加快電極的空穴向電解液中移動進而提高水氧化的效率,減少電極表面的空穴積累,從而抑制了電子-空穴復(fù)合并減少了CdS由于光生空穴過多產(chǎn)生的自身氧化。
4 結(jié)論
綜上,采用簡易SILAR法構(gòu)建了TiO2/CdS/Co-Pi光陽極,將其用于光催化水氧化,在中性的磷酸緩沖溶液中,在400 nm以上波長光照下,外加偏壓為0 V(vs Ag/AgCl)時得到一個很高的光電流響應(yīng)(1.3 mA·cm-2)。與未引入Co-Pi催化劑的TiO2/ CdS光陽極相比,光電流密度提高了25%,并且穩(wěn)定后仍能保持較高的光電流響應(yīng),表明以Co-Pi作為催化劑能極大地抑制光生電子和空穴的復(fù)合,加強光陽極的水氧化能力,進一步證實了TiO2/ CdS/Co-Pi體系通過可見光驅(qū)動分解水將太陽能轉(zhuǎn)化成化學(xué)能的可能性。
(1) McEvoy,J.P.;Brudvig,G.W.Chem.Rev.2006,106,4455. doi:10.1021/cr0204294
(2) Karkas,M.D.;Verho,O.;Johnston,E.V.;Akermark,B.Chem. Rev.2014,114,11863.doi:10.1021/cr400572f
(3) Fujishima,A.;Honda,K.Nature 1972,238,37.doi:10.1038/ 238037a0
(4) Chang,X.X.;Gong,J.L.Acta Phys.-Chim.Sin.2016,32(1),2 [常曉俠,鞏金龍.物理化學(xué)學(xué)報,2016,32(1),2.doi:10.3866/ PKU.WHXB201510192
(5) Ai,G.J.;Li,S.P.;Mo,R.;Zhong,J.X.Adv.Funct.Mater. 2015,25,5706.doi:10.1002/adfm.201502461
(6) Li,J.T.;Wu,N.Q.Catal.Sci.Technol.2015,5,1360. doi:10.1039/c4cy00974f
(7) Jia,H.M.;Xu,H.;Hu,Y.;Tang,Y.W.;Zhang,L.Z. Electrochem.Commun.2007,9,354.doi:10.1016/j. elecom.2006.10.010
(8) Sun,J.W.;Zhong,D.K.;Gamelin,D.R.Energy Environ.Sci. 2010,3,1252.doi:10.1039/C0EE00030B
(9) Cheng,C.W.;Zhang,H.F.;Ren,W.N.Nano Energy 2013,2, 779.doi:10.1016/j.nanoen.2013.01.010
(10) Xu,Y.;Schoonen,M.A.A.Am.Mineral.2000,85,543. doi:10.2138/am-2000-0416
(11) Kongkanand,A.;Tvrdy,K.;Takechi,K.;Kuno,M.;Kamat,P.V. J.Am.Chem.Soc.2008,130,4007.doi:10.1021/ja0782706
(12) Liang,Y.;Kong,B.;Zhu,A.W.;Wang,Z.;Tian,Y.Chem. Commun.2012,48,245.doi:10.1039/C1CC16060E
(13) Youngblood,W.J.;Lee,S.A.;Kobayashi,Y.;Hernandez-Pagan, E.A.;Hoertz,P.G.;Moore,T.A.;Moore,A.L.;Gust,D.; Mallouk,T.E.J.Am.Chem.Soc.2009,131,926.doi:10.1021/ ja809108y
(14) Xiang,X.;Fielden,J.;Huang,Z.Q.;Zhang,N.F.;Luo,Z.; Musaev,D.G.;Lian,T.Q.;Hill,C.J.Phys.Chem.C 2013,117, 918.doi:10.1021/jp312092u
(15) Xi,L.F.;Tran,P.D.;Chiam,S.Y.;Loo,J.S.C.;Wong,L.H. J.Phys.Chem.C 2012,116,13884.doi:10.1021/jp304285r
(16) Respinis,M.;Joya,K.S.;Groot,H.J.M.D.;Smith,W.A.; Krol,R.;Dam,B.J.Phys.Chem.C 2015,119,7275. doi:10.1021/acs.jpcc.5b00287
(17) Wang,L.;Dionigi,F.;Nguyen,N.T.;Kirchgeorg,R.;Gliech, M.;Grigorescu,S.;Strasser,P.;Schmuki,P.Chem.Mater.2015, 27,2360.doi:10.1021/cm503887t
(18) Kanan,M.W.;Nocera,D.G.Science 2008,321,1072. doi:10.1126/science.1162018
(19) Zhong,D.K.;Sun,J.;Inumaru,H.;Gamelin,D.R.J.Am. Chem.Soc.2009,131,6086.doi:10.1021/ja9016478
(20) Seabold,J.A.;Choi,K.S.Chem.Mater.2011,23,1105. doi:10.1021/cm1019469
(21) Li,X.R.;Wang,J.G.;Men,Y.;Bian,Z.F.Applied Catalysis B: Environmental.2016,187,115.doi:10.1016/j. apcatb.2016.01.034
(22) Bjelajac,A.;Djokic,V.;Petrovic,R.;Socol,G.;Mihailescuc,I. N.;Florea,I.;Ersend,O.;Janackovic,D.Applied Surface Science 2014,309,225.doi:10.1016/j.apsusc.2014.05.015
(23) Pan,Y.X.;Zhou,T.H.;Han,J.Y.;Hong,J.D.;Wang,Y.B.; Zhang,W.;Xu,R.Catal.Sci.Technol.2016,6,2206. doi:10.1039/c5cy01634g
(24) Na,Y.;Hu,B.;Yang,Y.L.;Zhou,L.Chin.Chem.Lett.2015, 26,141.doi:10.1016/j.cclet.2014.09.011
(25) Du,P.W.;Kokhan,O.;Chapman,K.W.;Chupas,P.J.;Tiede, D.M.J.Am.Chem.Soc.2012,134,11096.doi:10.1021/ ja303826a
(26) Farrow,C.L.;Bediako,D.K.;Surendranath,Y.;Nocera,D.G.; Billinge,S.J.L.J.Am.Chem.Soc.2013,135,6403. doi:10.1021/ja401276f
(27) Costentin,C.;Porter,T.R.;Saveant,J.M.J.Am.Chem.Soc. 2016,138,5615.doi:10.1021/jacs.6b00737
(28) Cobo,S.;Heidkamp,J.;Fize,J.;Fourmond,V.;Guetaz,L.; Jousselme,B.;Ivanova,V.;Dau,H.;Fontecave,M.;Artero,V.; Fontecave,M.;Artero,V.Nat.Mater.2012,11,802. doi:10.1038/nmat3385
Preparation and Performance of a SILAR TiO2/CdS/Co-Pi Water Oxidation Photoanode
ZHOU Li*LIU Huan-Huan YANG Yu-Lin QIANG Liang-Sheng
(School of Chemistry and Chemical Engineering,Harbin Institute of Technology,Harbin 150001,P.R.China)
Asemiconductor heterostructure of TiO2/CdS/cobalt phosphate water oxidation catalyst(Co-Pi WOC) photoanode was fabricated by the successive ionic layer adsorption and reaction(SILAR)procedure and photoassisted electro-deposition.The structure,morphologys and magnetic properties of the resultant particles were characterized using X-ray diffraction(XRD),scanning electron microscope(SEM),and X-ray photoelectron spectroscopy(XPS).CdS and Co-Pi quantum dots loaded on to the TiO2nanofilm.The TiO2/CdS/Co-Pi photoanode had an initial photocurrent of 1.3 mA·cm-2and a stable level of 0.5 mA·cm-2.A relatively stable level was maintained under visible light irradiation in neutral solution,especially at the low bias voltage of 0 V (vs Ag/AgCl).In this system,CdS quantum dots serve as the light absorber and generate electron holes;the Co-Pi WOC acts as a hole transfer layer that can transfer the hole for water oxidation;and the TiO2is the electron conductor for efficient charge transfer to the cathode to actualize proton reduction.
Photocatalysis;Water oxidation;Successive ionic layer adsorption and reaction;Visible light;TiO2/CdS/Co-Pi photoanode
O644
10.3866/PKU.WHXB201608232
Received:May 31,2016;Revised:August 15,2016;Published online:August 23,2016.
*Corresponding author.Email:lizhou@hit.edu.cn;Tel:+86-451-86413702-805.
The project was supported by the Heilongjiang Postdoctoral Fund,China(LBH-Z14106)and Program for Innovation Research of Science in Harbin Institute of Technology,China(Q201508).
黑龍江省博士后經(jīng)費(LBH-Z14106)和哈爾濱工業(yè)大學(xué)理學(xué)創(chuàng)新研究發(fā)展培育計劃(HIT Q201508)資助
