3C-like蛋白酶抑制劑的構效關系、分子對接和分子動力學
2016-12-29 05:42:39付新梅蔣思宇王景輝張述偉
物理化學學報 2016年11期
關鍵詞:模型
林 峰 付新梅 王 超 蔣思宇 王景輝張述偉 楊 凌 李 燕,*
(1大連理工大學化工學院,遼寧大連116024;2大連理工大學精細化工國家重點實驗室,遼寧大連116024;3中國科學院大連化學物理研究所,藥物資源開發研究組,遼寧大連116023)
3C-like蛋白酶抑制劑的構效關系、分子對接和分子動力學
林 峰1付新梅2,*王 超1蔣思宇1王景輝1張述偉1楊 凌3李 燕1,*
(1大連理工大學化工學院,遼寧大連116024;2大連理工大學精細化工國家重點實驗室,遼寧大連116024;3中國科學院大連化學物理研究所,藥物資源開發研究組,遼寧大連116023)
3C-like蛋白酶是中東呼吸綜合征冠狀病毒(MERS-CoV)等其它冠狀病毒的繁殖過程中極為重要的蛋白酶。它已成為人類在抗冠狀病毒領域中的研究熱點。本文基于計算生物學方法對與MERS-CoV同屬的蝙蝠冠狀病毒HKU4(HKU4-CoV)的43個肽類3C-like蛋白酶抑制劑分子,建立三維定量構效關系(3D-QSAR)模型。在基于配體疊合的基礎上,發現比較分子相似性指數分析法(CoMSIA)中的四個場組合(位阻場、靜電場、氫鍵供體場與氫鍵受體場)為最優的模型(Q2=0.522,R2ncv=0.996,R2pre=0.904;Q2:交叉驗證相關系數,R2ncv:非交叉驗證相關系數,R2pre:驗證集分子的預測值相關系數),并借助該模型通過分子對接(docking)與分子動力學(MD)方法闡明了配受體結合作用。實驗結果表明:(1)基于最優的CoMSIA模型基礎上的三維等勢圖形象地說明了分子基團的位阻作用、靜電作用、氫鍵供體與氫鍵受體作用對分子生物活性的影響;(2)分子對接研究結果顯示了疏水性以及結晶水、氨基酸His166和Glu169在配體和受體結合過程中產生重要作用;(3)分子動力學模擬進一步驗證了分子對接模型的可靠性,并發現了兩個新的關鍵氨基酸Ser24與Gln192,它們與配體產生了兩個較強的氫鍵。此外,根據這些結果,一些新的具有潛在抑制活性的肽類化合物作為3C-like蛋白酶抑制劑被獲得。以上結果能夠幫助深入了解3C-like蛋白酶與肽類抑制劑的作用機理,并且能夠為今后的抗MERS-CoV藥物設計提供有價值的參考。
中東呼吸綜合征冠狀病毒;3C-like蛋白酶;肽類抑制劑;三維定量構效關系;分子對接;分子動力學
1 引言
冠狀病毒(CoV)是一類分布廣泛的,具有重大威脅的病原體1,2。該種病毒能引起人及哺乳類動物疾病,且被感染的人或動物可能會成為呼吸道、腸道、肝臟、神經系統疾病的攜帶者。最近出現的中東呼吸綜合征冠狀病毒(MERS-CoV)就是其中能感染人類并傳播的冠狀病毒之一3。該病毒于2012年9月在中東地區首次被鑒定出來,隨后逐漸擴散,在很多國家都報道了感染病例。MERS-CoV能導致非常嚴重的呼吸道病癥,致死率高。截至2015年1月30日,共有956例確診病例,死亡351例,死亡率高達36.7%,遠高于嚴重急性呼吸綜合征冠狀病毒(SARS-CoV),引發了全球關于MERS大流行潛在可能性的恐慌4。因此,MERS-CoV已經成為一種全球范圍內嚴重威脅人類生命安全的新型冠狀病毒。
冠狀病毒被國際病毒分類委員會(ICTV)分為四個屬,即α、β、γ和δ冠狀病毒屬。其中,β冠狀病毒屬有四個亞群(A-D),通過將MERS-CoV和其他β冠狀病毒基因序列對比發現,MERS-CoV與SARS-CoV中較保守的核酸復制酶序列只有小于50%的氨基酸相同,而與從蝙蝠體內分離到的蝙蝠冠狀病毒HKU4(HKU4-CoV)和HKU5 (HKU5-CoV)親緣關系很近,相似度分別約為75%和76.7%5-7。因此,ICTV在β冠狀病毒屬C亞群中增加MERS-CoV為一個新種,它也是該亞群中第一種能夠感染人類的β冠狀病毒。研究表明,MERS-CoV不僅可存在于蝙蝠體內,其也被發現能生存在卡塔爾單峰駱駝體內,而且它們的輔助蛋白在體外能夠抑制人類抗病毒的信號路徑3,8-10。除此之外,Wang等11通過研究認為MERS-CoV的起源是HKU4-CoV或HKU5-CoV的變異,其中更多研究表明,其更傾向是來自HKU4-CoV的變異,表明從蝙蝠到人類的一種人畜共患的轉變。
3C-like蛋白酶(3CLpro)是病毒繁殖的蛋白水解過程中,兩個非常重要的病毒半胱氨酸蛋白酶之一12-14。3C-like蛋白酶在擁有典型的Leu-Gln (Ser、Ala、Gly)氨基酸序列的11個保留位點上將復制酶多聚蛋白斷裂,其中它們的P1位置與P2位置分別擁有完整的Gln氨基酸殘基和脂肪族氨基酸殘基15,16。迄今為止,沒有疫苗和抗病毒藥物能夠真正有效預防冠狀病毒感染人類或治療已患病的人類。因此,3C-like蛋白酶對于冠狀病毒生命周期起著至關重要的作用,使其成為一個抗MERSCoV藥物的發展很有吸引力的目標,基于3C-like蛋白酶抑制劑的研究具有廣闊的前景17-19。
隨著計算機科技的高速發展,三維定量構效關系(3D-QSAR)作為一個十分有效、經濟的方法,已被廣泛應用于各個領域的結構與活性、結構與性質關系的研究,來幫助設計新的藥物20,21。3DQSAR包括比較分子力場分析(CoMFA)和在其基礎之上發展起來的較新穎的比較分子相似性指數分析(CoMSIA)等眾多方法。這些方法認為,配體與受體之間的相互作用取決于化合物周圍分子力場的差異,以定量化的分子力場參數作為變量,對藥物活性進行回歸分析便可反映出藥物小分子與生物大分子之間的作用模式,進而有選擇地設計新藥。
目前MERS-CoV的3C-like蛋白酶抑制劑還處于研究階段,但已有文獻證明與MERS-CoV同屬的SARS-CoV的3C-like蛋白酶抑制劑也同樣能良好地抑制MERS-CoV的繁殖22-25。本文通過上述計算方法,對新合成的與MERS-CoV更接近的HKU4-CoV的43個肽類3C-like蛋白酶抑制劑分子進行了研究,建立了3D-QSAR模型,并探討了此類抑制劑周圍的位阻場、靜電場、氫鍵供體場和氫鍵受體場對生物活性的影響。此外,我們還利用分子對接方法來驗證3D-QSAR模型的準確性和穩定性,之后采用分子動力學方法對分子對接的結果進行補充。它們能幫助我們更好地預測抑制劑小分子與靶點蛋白大分子之間可能的結合構象,進而預測小分子與蛋白的結合力和其生物活性。我們希望獲得的分子構效關系以及配受體結合作用的結論能夠延伸到為今后抗MERS-CoV藥物(3C-like蛋白酶抑制劑)的設計提供理論指導。
2 實驗部分
2.1數據庫及生理活性
本文模型的建立及分析采用的是Tripos公司的SYBYL 6.9設計軟件包。文中所研究具有抑制3C-like蛋白酶作用的肽類化合物結構和活性數據均來自于文獻26。這些分子的抑制活性范圍IC50較寬,將其值轉化pIC50(-logIC50)值作為定量構效關系分析中的因變量,使數值均勻地分布在4.25-6.50之間。整個數據集以近似3:1的比例,被分為兩個部分。其中,33個分子結構和活性作為訓練集,用于模型的構建;而其余10個分子則加入到驗證集中,用于測試該模型的準確性。訓練集和驗證集的選取應遵循隨機挑選的原則,在盡可能滿足更好的結構差異性與多樣性的同時,使訓練集與驗證集的分子比較均勻地分散在整個數據庫范圍中。我們將所有的分子結構及其對應的生理活性pIC50實驗值列于表S1(Supporting Information)中。其中,表S1還包含基于配體和受體的CoMFA與CoMSIA模型得到的pIC50預測值,表S1粗體字顯示的部分是具有最佳預測性能的模型的pIC50預測值。
2.2構象優化及分子疊合
在建立3D-QSAR模型中,分子疊合是十分重要的環節,其直接決定了模型的優劣,而在分子疊合前需要進行構象優化27。分子三維構象優化的步驟如下:
(1)采用Chemoffice軟件構建化合物分子的二維和三維結構;
(2)導入到SYBYL中進行能量優化,每個分子上加載Gasteiger-Hückel電荷28;
(3)利用Tripos力場29作為能量最小化和構象搜索的共軛方法,且收斂條件設置為0.00419 kJ· mol-1;
(4)采用Powell能量梯度獲得穩定構象,且最大優化次數設置為1000次,梯度設置為2.095 kJ· mol-1·nm-1。
在整個數據集中,我們選擇活性最大的1號分子進行疊合,應用SYBYL軟件,在1號分子上劃定所有分子的分子骨架,用紫色線顯示,如圖1a所示。在研究中,使用了三種不同的疊合方式:疊合I是基于配體的疊合,得到疊合圖為圖1b;疊合II是基于受體完成的,將所有分子都對接于受體中,得到對接結果最高打分的構象進行疊合,分子疊合圖為圖1c;疊合III也是一種基于受體的疊合方法,其中所有的分子構象均來自于疊合II,隨后經歷疊合I的疊合過程,并得到疊合圖為圖1d。
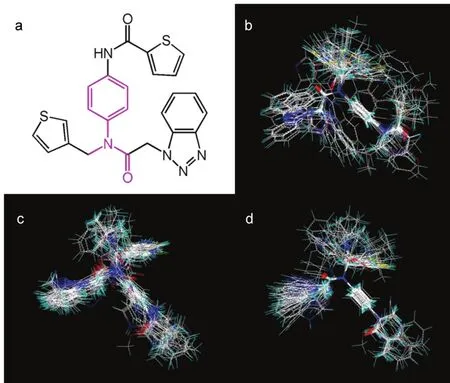
圖1 模板分子結構及三種分子疊合圖Fig.1 Structure of template molecule and three kinds of molecular alignment
2.3CoMFA及CoMSIA研究
CoMFA及CoMSIA都是應用最廣泛的合理藥物設計方法。CoMFA基于的假設是配體與受體的相關作用是非共價性的相互作用,利用分子力學力場(位阻場和靜電場)可以較好地解釋它們的相關作用30。相比之下,CoMSIA是以CoMFA為基礎,并對其進行了補充和優化。CoMSIA將CoMFA中的位阻場和靜電場進一步細分為五個場,即多引入了疏水、氫鍵供體和氫鍵受體場,所以更細致地描述了配體與受體間的相互作用。此外,CoMSIA改變了探針原子與配體之間作用能的計算公式,非CoMFA傳統的庫侖(Coulomb)和范德華(Lennard-Jones)函數,而采用了更加平滑的高斯(Gaussian)函數來計算分子場的數據,從而有效避免了原子位置發生異常和配體周圍不同探針位點上勢能的差異31。而且在CoMSIA中,分子力場能量于不同的探針位點間衰減很快,致使計算過程自動收斂,無需再設定能量截斷值。
在本文的研究中,我們采用了CoMFA和CoMSIA兩種定量構效關系方法,用來間接地反映了化合物分子與受體相互作用過程中兩者之間的非鍵相互作用特征,并預測良好的三維定量構效關系模型。分子疊合后,分子空間取向基本一致,然后用一個粒子探針在分子周圍的空間中游走,計算粒子探針與分子之間的相互作用,并記錄下在空間不同坐標中它們相互作用的能量值,進而獲得分子力場數據。在CoMFA建模方法中,用sp3雜化的C+為探針,探針所帶電荷為+1.0e,半徑為0.152 nm,默認步長為0.2 nm,對整個三維網格進行搜索,且位阻場和靜電場效應能的閾值分別設定為默認值209.29 kJ·mol-1,同時其它參數均為默認值。對于CoMSIA建模中,用到的三維網格和CoMFA相同,以sp3雜化的C+為探針,電荷為+1.0e,半徑為0.100 nm,疏水性為+1.0,氫鍵供體和受體強度均為+1.0。此外,構建這兩種的QSAR模型時,衰減因子均設定為默認值0.3。
2.4PLS分析與驗證
本文采用偏最小二乘回歸分析(PLS)來建立CoMFA/CoMSIA描述符和抑制劑pIC50值之間的關系。在PLS的建模中,為了避免過度擬合問題,必須要對其中每個連續組分的重要性進行嚴格測試,然后測試會在組分不重要時停止。交叉驗證是一種能測試這種重要性的實用且可靠的方法32。我們運用的交叉驗證方法是抽一法(leave-oneout),它是依次從訓練集中抽取一個樣本,余下的n-1個樣本作為訓練集構建模型,并用該模型預測抽取出的樣本,直到每個分子都被取出預測一次,最終可以得到最佳組分數(OPN),與能夠衡量所建模型的內部預測能力的重要統計指標,交叉驗證相關系數(Q2)。而利用所有訓練集的分子結合OPN,通過非交叉驗證分析計算可以得出非交叉驗證相關系數(R2ncv)、統計值(F)、訓練集的估計標準誤差(SEE)和驗證集的最小標準預測偏差(SEP)33。此外,根據預測驗證集的活性,我們還可以得到能夠評估模型的外部預測能力的重要參數,驗證集分子的預測值相關系數(R2pre)。因此采用PLS能在避免過擬合現象和消除大多數可變因素的同時,建立大量的QSAR方程34,35。最后,我們將CoMFA/CoMSIA結果以等勢圖的形式直觀地表示出來,并進行下一步的分析。
2.5分子對接
為揭示肽類抑制劑小分子和蛋白大分子之間的結合機制,以及確定抑制劑的生物活性構型,我們從Protein Data Bank數據庫36中選擇蛋白靶點的三維晶體結構PDB ID:4YOI26,基于遺傳算法的GOLD 5.0版本軟件37,進行分子對接研究。分子對接是利用配體小分子與受體蛋白大分子的完整結構信息,計算把配體放在受體活性位點的位置,然后按照能量匹配、幾何匹配以及化學環境匹配的原則來評價抑制劑和蛋白的結合能力,并預測它們之間最佳的結合模式38。在實際過程中,不僅配體小分子具有柔性易產生多種構象,而且受體大分子也是柔性且構象可發生變化。在兩者分子對接過程中,它們的構象會變化以找到一個能達成局部穩定的契合點。因此,研究人員可以通過分子對接確定柔性的配體與受體蛋白的結合位置和空間取向,以此來研究藥物的作用機制并指導新藥物的設計。
分子對接之前,先對蛋白質模型進行修改,只留下單鏈,并在蛋白質結構中加入極性氫,且刪除原有的配體與雜原子。我們在GOLD中將受體大分子中每一個原子坐標1 nm空間內的氨基酸殘基所組成的空間范圍設為可能與配體小分子的結合位點。我們將43個分子分別對接到潛在的結合位點中,每次對接都會產生5個左右可能的構象。所有構象都會經過GOLD評估,對其適當性進行打分。打分的分值計算考慮到了配體分子內氫鍵和拉力的作用,以及配體與受體之間相互的氫鍵和范德華力的貢獻39。在分子對接過程中,打分函數采用GoldScore算法,其他參數采用軟件中的默認值。
2.6分子動力學
為了進一步檢驗分子對接研究中結果的穩定性,以及構建一個能夠表現分子所在真實環境的蛋白質模型,即需要考慮蛋白質大分子的動態靈活性,我們應用GROMACS軟件包中的GROMOS96力場40,41對分子對接得到的復合物開展了詳盡的分子動力學模擬。分子動力學模擬是研究生物大分子體系的常用計算方法。該法根據牛頓經典力學原理,模擬分子體系的運動狀態,計算在相空間中所有原子的運動軌跡,接著在從各種狀態的分子體系構成的系綜中,選取樣本計算構型積分,且以此為基礎,進行計算該體系的熱力學量等其他宏觀性質。
進行動力學模擬之前,需要對模擬體系進行一系列預處理。我們在PRODRG 2.542中獲得配體-蛋白質復合物分子的拓撲文件,隨后將初始復合物放置在一個邊長為10 nm的周期性立方體晶格體中,其中保持晶格上每一點與蛋白質的距離至少是1 nm41。為了保證體系的電中性,蛋白質復合物和水中所有原子均被隨機地放置于晶格盒子中,并用簡單點電荷水來填充模擬體系的其余部分43。詳細的動力學模擬步驟如下:首先為了避免分子間高能碰撞的可能性,對整個系統進行2500步最快下降法操作,令其能量逐步最小化,再進行2500步共軛梯度優化。接著,當整個系統的溫度和壓力上升到300 K和101.325 kPa且處于每納米8.38 kJ·mol-1穩定力時,對常溫常壓NPT系綜進行50 ps恒壓力與500 ps密度平衡操作。最后,在周期性邊界條件下,設定積分步長為0.002 ps,在保持恒溫的條件下進行50 ns動力學模擬。在整個模擬過程中,我們分別采用了SHAKE算法44與PME方法45來固定含碳氫鍵和處理庫侖作用。
3 結果與討論
3.1 CoMFA與CoMSIA模擬統計量分析
在之前的研究工作中,我們分別運用了基于配體和基于受體的三種疊合方式對數據集中43個肽類分子進行疊合,生成了六個不同的疊合模型,并對CoMFA兩種力場(位阻場與靜電場)和CoMSIA五種力場(位阻場、靜電場、疏水場、氫鍵供體場和氫鍵受體場)的所有3種與31種排列組合進行了計算。為了提高模型的可對比性,所有的CoMFA和CoMSIA模型在建模過程中都使用相同的訓練集(33個分子)和驗證集(10個分子)。通過對比模型結果,得出基于配體得到的模型最優,因此我們主要分析基于配體的模型結果。這些模型的統計參數列于表1中。
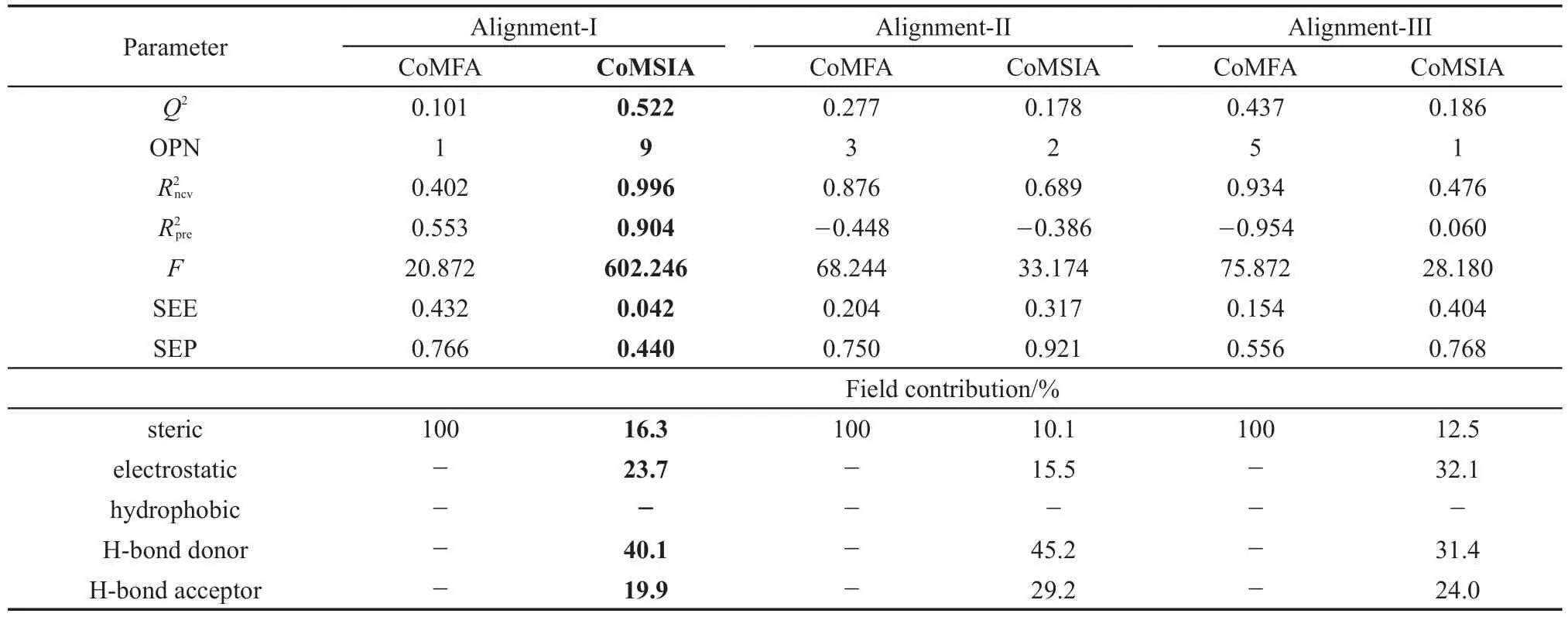
表13D-QSAR結果匯總Table 1 Summary of 3D-QSAR results
對比得到了一個具有最佳預測性能的模型結果(表1中黑體字顯示),并用PLS法和抽一法對其進行評估,獲得包括用來檢驗所建模型是否有效的3個參數(Q2、OPN與R2ncv)和用來檢驗模型預測能力的驗證集R2pre。此外,還包括一些其他的統計參數,如模型標準誤差,即F值、訓練集的SEE和驗證集的SEP,以及各個力場對模型的貢獻率。
從統計意義上來說,一個良好的3D-QSAR模型,前提必須滿足四個重要條件:Q2>0.5且,并有較低的SEE、較大的F值,才能很好地預示CoMFA和CoMSIA模型46。因此我們通過建模分析,最終以組合了擁有最高的的位阻場、靜電場、氫鍵供體場和氫鍵受體場的CoMSIA模型為最優模型。其Q2=0.522、OPN= 9、R2ncv=0.996、F=602.246和SEE=0.042,可以看出這個模型的誤差較小且可靠性較高。當被驗證集驗證時,所得模型R2pre=0.904與SEP=0.440,展示出其良好的預測能力。所有這些數據結果都說明了我們所建立的基于配體疊合的CoMSIA模型是一個可靠的具有很好預測能力的模型。
同時,圖2中顯示了在最優的CoMSIA模型中,所有訓練集分子和驗證集分子的預測pIC50值和實際pIC50值的散點圖。其中訓練集分子以方塊代表,而驗證集分子以三角表示。從圖中可以看出,整個數據集都是密集地分布在回歸線的兩側,而且抑制劑于模型中預測的pIC50值與實驗中測得的pIC50值的差值較小。這表明訓練集和驗證集的密集度和誤差都很相似,未出現過擬合時訓練集分布均勻,而驗證集誤差較大(離回歸線很遠)的問題,且表明預測值和實際值的相關性良好,得到了比較可靠的構效關系模型47,48。
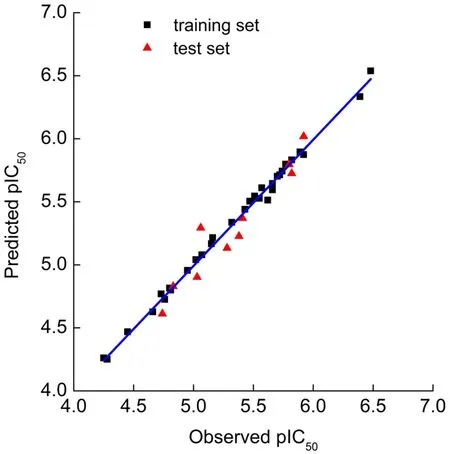
圖2 基于配體的CoMSIA模型的預測與實際pIC50值的關聯圖Fig.2 Ligand-based correlation plot of the predicted versus the actual pIC50values based on the CoMSIAmodel
3.23D-QSAR等勢圖結果與分析
為進一步形象化和具體化得到的CoMSIA模型中包含的信息和內容,我們分析CoMSIA模型中四個力場的等勢圖。這些等勢圖能夠直觀地反映抑制劑小分子的生物活性與其化學結構上的相互聯系,從而探究出對藥物分子活性有關的重要因素,進而在實際藥物設計中提供指導49。在繪制等勢圖過程中,為了能夠更好地分析各種等勢圖的結果,一致將活性最高的分子,1號分子(pIC50= 6.481)選為模板分子,疊放于四張等勢圖(圖3)中,以便于對比。在該1號分子的三維結構(圖4)中,R1、R2、R3和R4取代基是數據集內其他分子結構與1號分子的差別所在。此外,在該CoMSIA模型四個力場的等勢圖中,積極影響區域和消極影響區域貢獻比率分別設置為默認值80%和20%。
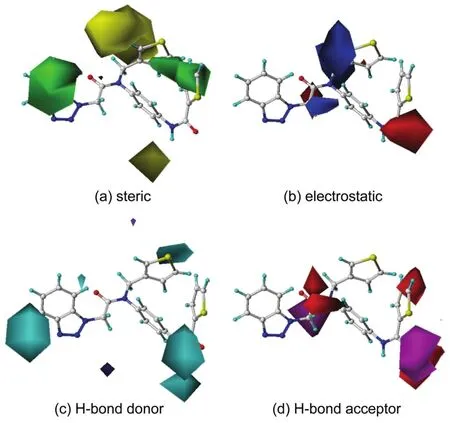
圖3 結合1號分子的CoMSIA模型等勢圖Fig.3 CoMSIAcontour maps in combination with compound 1
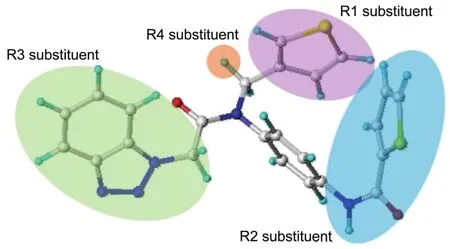
圖4 1號分子結構Fig.4 Structure of compound 1
圖3(a)為基于1號分子CoMSIA位阻場的等勢圖。其中,綠色和黃色云團分別表示位阻基團對分子活性的有利和有害區域。由圖可看出,R3取代基遠離分子骨架的部分和遠處R1與R2取代基之間有很大的綠色區,說明R3取代基遠位以及R1與R2取代基相近部分存在的較大基團有益于提高分子活性,這一點從數據集中得到了充分的驗證。例如,24號分子(pIC50=5.658)與34號分子(pIC50= 4.954)對比,以及1號分子(pIC50=6.481)與13號分子(pIC50=5.509)對比,它們的差別只在對應位置存在較大基團,使位阻變大,有益于提高抑制活性。此外,R2取代基靠近分子骨架苯環的部分和R4取代基周圍存在兩塊黃色云團,顯示了位阻較大基團在這片區域對分子活性起抑制作用。例如2號分子(pIC50=6.387)和28號分子(pIC50=5.377)的R4取代基中,H原子的位阻明顯小于叔丁基的位阻,使得28號分子較2號分子活性低很多。
圖3(b)為基于1號分子CoMSIA靜電場的等勢圖。其中,藍色云團代表正電性積極影響區域;而紅色云團表示負電性積極影響區域。圖中R4取代基臨近分子骨架的部分有大片藍色區域,說明此處正電性取代基的存在對提高抑制活性有利,如在此處帶有正電性的叔丁基基團的分子活性較大。而圖中R2取代基臨近分子骨架的部分有大片紅色區域,另外還有小片紅色區域在R3取代基靠近分子骨架氧原子的部分,說明負電性取代基在此處有利。圖可見,R2取代基臨近分子骨架部分和R1與R3取代基遠離分子骨架部分附近有較大的青色區域,說明該區域有氫鍵供體作用的取代基對提高分子活性有利。例如,最高活性的1號分子在R2取代基處有N―取代基(其上擁有氫原子作為氫鍵供體),其抑制活性遠遠高于其它無氫鍵供體基團或N―取代基(其上未擁有氫原子)的類似物分子,即是一很好的佐證。總的來說此處氫鍵供體基團能提高抑制劑活性。同時,R3與R4取代基的遠位有兩個極小紫色云團,其對分子活性具有一定消極影響,但從大趨勢來看,基本可以忽略。
圖3(d)為基于1號分子CoMSIA氫鍵受體場的等勢圖。其中,紫色云團代表更依賴氫鍵受體區域,氫鍵受體在此處更益于生物活性,而紅色云團則相反。從圖可見,紫色與紅色云團基本都同時出現在R2與R3取代基靠近分子骨架部分,但它們都處于非同側,此外還有一塊紅色云團在R2取代基離骨架的遠位。這使分子具有一定的敏感性,說明R2與R3取代基的空間旋轉角度不同,導致其受氫鍵受體場的影響有巨大差異。當R2與R3取代基能作為氫鍵的接收基團時,且接收基團都恰好旋轉到空間中氫鍵受體有益活性區域時,積極作用達到最大;反之,消極作用最大。例如,在基于9號分子(pIC50=5.699)的CoMSIA氫鍵受體場的等勢圖(圖5)中,可以看到,9號分子的R2取代基上具有一個S原子和兩個N原子可作為氫鍵受體,R2取代基的角度已基本達到使它們靠近紫色積極云團而遠離紅色云團;同樣,該現象也可從這個分子的R3取代基的三個N原子上看到。氫鍵受體場對9號分子的積極作用最大,這可能是證明該分子在數據集中具有較高活性的原因之一。
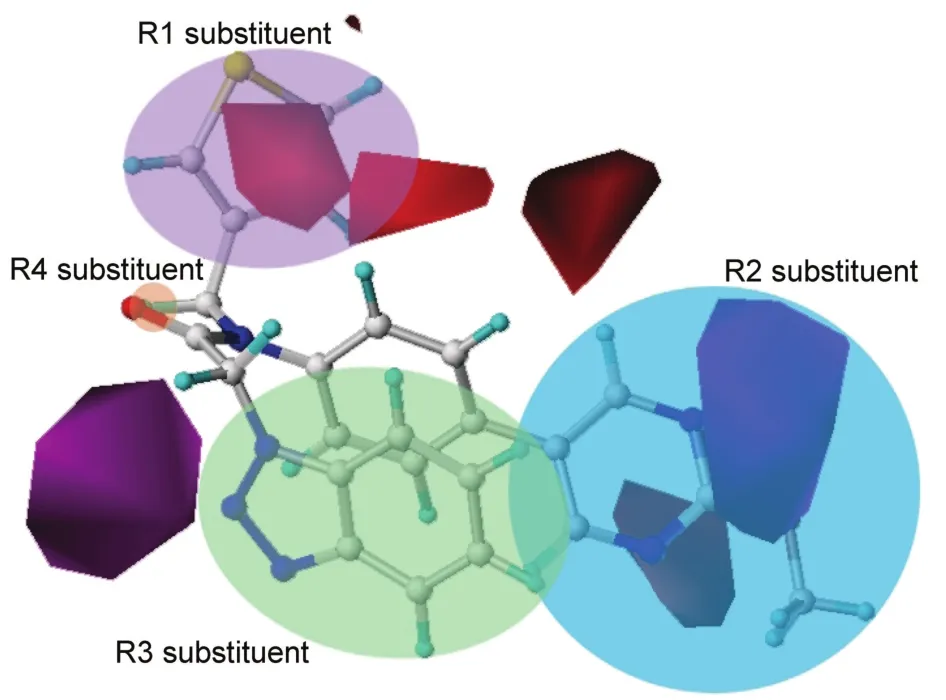
圖5 結合9號分子的CoMSIA模型氫鍵受體輪廓圖Fig.5 CoMSIAH-bond acceptor contour map in combination with compound 9
綜合以上3D-QSAR的研究結果,我們分析了影響分子抑制活性的各基團作用域(圖6)。我們能夠得出概括性的關于肽類分子的結構性優化方面的結論:
(1)對于R1取代基,遠離分子骨架的部分(靠近R2取代基的周圍),如圖6紅色區域內,大位阻基團和氫鍵供體基團有利于分子活性的提高;
(2)對于R2取代基,臨近分子骨架的部分,如圖6深藍色區域內,負電性基團、氫鍵供體基團和氫鍵受體基團有利于分子活性的提高;此外,靠近R1取代基的周圍,如圖6淺藍色區域內,大位阻基團有利于分子活性的提高;
(3)對于R3取代基,臨近分子骨架的部分,如圖6深綠色區域內,負電性基團和氫鍵受體基團有利于分子活性的提高;此外,遠離分子骨架的部分,如圖6淺綠色區域內,大位阻基團和氫鍵供體基團有利于分子活性的提高;
(4)對于R4取代基,臨近分子骨架的部分,如圖6黃色區域內,正電性取代基有利于分子活性的提高。
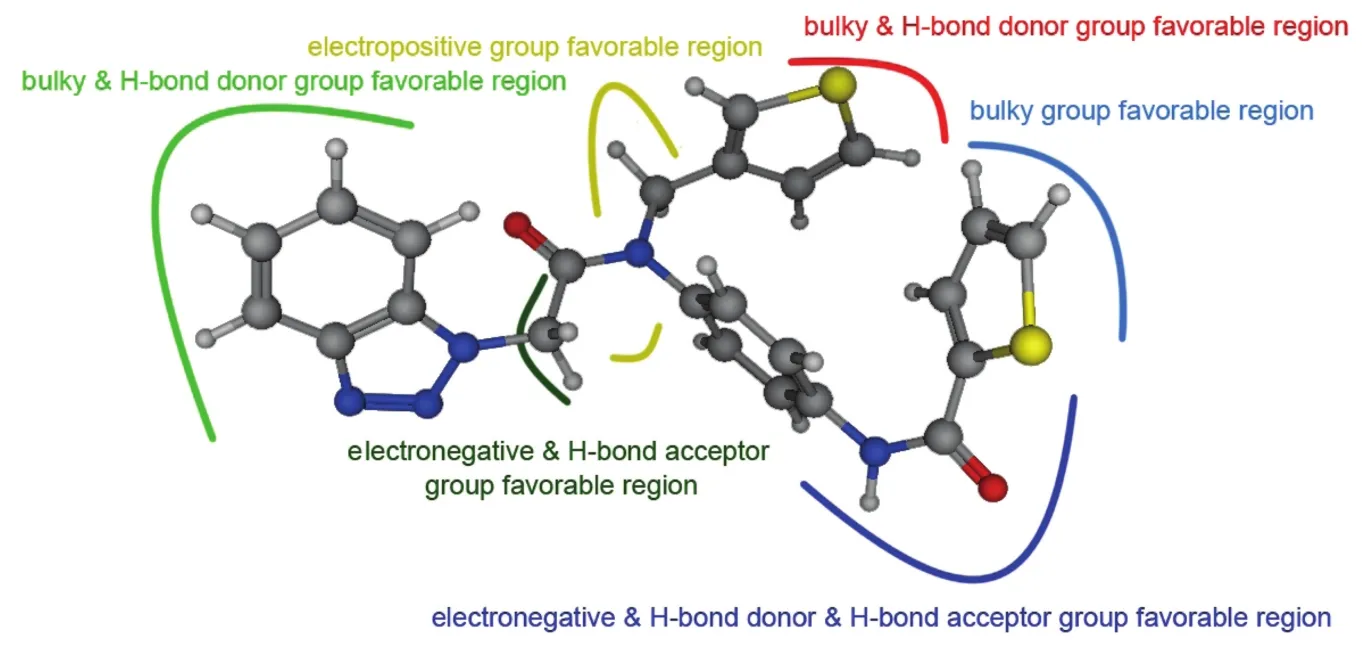
圖6 分子抑制作用的交互特性Fig.6 Interaction features of compound impacting the inhibitory effect color online
3.3分子對接研究
分子對接是一種用于驗證3D-QSAR模型準確性和穩定性的有效工具。它能幫助我們更好地預測候選藥物小分子與靶點蛋白之間可能的結合構象,進而預測小分子的結合力和生物活性。在本文的研究中,為了闡明所選肽類3C-like蛋白酶抑制劑分子能否調節靶點蛋白,同時為了進一步形象化地展現它們之間的作用機制及結合構象,我們對整個數據集中所有43個肽類分子都進行了對接研究。
為了之后能與John等26已做的X射線晶體實驗結果作對比分析,我們采用相同的條件(存在結晶水)的受體和配體分子做了分子對接。另外,我們也對比了不存在結晶水的受體和配體分子的對接結果。在它們的對接結果中,獲得得分最高的合理性打分,其平均分分別為81.519分與78.922分。從打分結果來看,與存在結晶水的對接結果相比,沒有結晶水的對接打分變低了2.597分,受體存在結晶水的對接效果確實比較好,驗證了John等26實驗條件的合理性。通過對存在結晶水的對接結果分析得到了結晶水HOH523與配體分子產生的氫鍵距離圖(圖7),其中它們的氫鍵距離范圍從0.234至0.334 nm。一般來說,形成氫鍵的幾何標準是距離小于0.35 nm,且它們之間所連成的角度必須大于120°,此外0.32-0.40 nm之間的距離可以稱為是弱氫鍵50。因此,HOH523能與大部分的配體(30/43)產生較強的氫鍵作用,表明了由于它們的氫鍵存在使配體更加穩定的結合于空腔中,進而影響到整體打分。最終,我們選擇1號配體分子與存在結晶水的受體大分子進行分子對接獲得的分數最高的、最合理的結果來進行接下來的分析。
圖8為1號分子和原來蛋白質晶體中結合的分子互相疊合的結果,它們分別呈橙色和綠色。從圖中可明顯看出,兩個分子在受體中的位置極其相似,這有力地證明了分子對接結果的準確性,能夠再現3C-like蛋白酶與其抑制劑分子間的實驗結合空腔。
在配體藥物分子與受體蛋白相結合時,其主要作用一般是與其周圍的氨基酸形成的疏水、氫鍵等相關作用。圖9展示了最高活性的1號分子在其周圍0.45 nm內的蛋白質空腔中的氨基酸殘基的對接作用模式,包含的氨基酸從圖中可以看出,抑制劑分子穩定在蛋白質空腔內,形成一個穩定的空間區域。從圖中配體分子周圍的氨基酸類型判斷,該分子結合的空腔有兩個疏水位點(文中標為S1和S2)。S1位點由氨基酸Ser24、Met25、ILe42、Pro45、Ala46以及Leu49等氨基酸構成。1號抑制劑分子的R2取代基部分正結合于此。S1位點中的氨基酸大部分都具有脂肪族氨基酸的特性,它們共同促進該疏水位點的形成。同時,1號抑制劑分子的R3取代基部分也被一個疏水性的S2位點所固定。這些氨基酸包括Leu27、Phe143、Leu144、Cys145、Met165、His166、Met168以及Glu169等,它們也有較強的脂肪特性使得配體分子能夠緊密固定在這個空腔中。
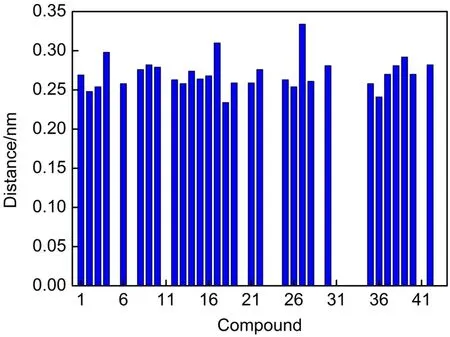
圖7 結晶水HOH523與配體分子的氫鍵距離Fig.7 H-bond lengths between HOH523 and ligand molecules
此外,以上對接結果圖顯示,配體還通過不同方向的四個氫鍵緊密地結合在受體空腔內。四個氫鍵分別為:
(1)配體分子R2取代基上的氮原子與結晶水HOH523氧原子形成的氫鍵(N―H…O,0.269 nm);
(2)配體分子R3取代基上的遠離骨架部分的氮原子與氨基酸His166側鏈上氮原子形成的氫鍵(N…H―N,0.290 nm);
(3)配體分子骨架上的氧原子與氨基酸Glu169骨架上氮原子形成的弱氫鍵(O…H―N,0.359 nm);
(4)配體分子R3取代基上的靠近骨架部分的氮原子與氨基酸Glu169骨架上氮原子形成的弱氫鍵(N…H―N,0.352 nm)。
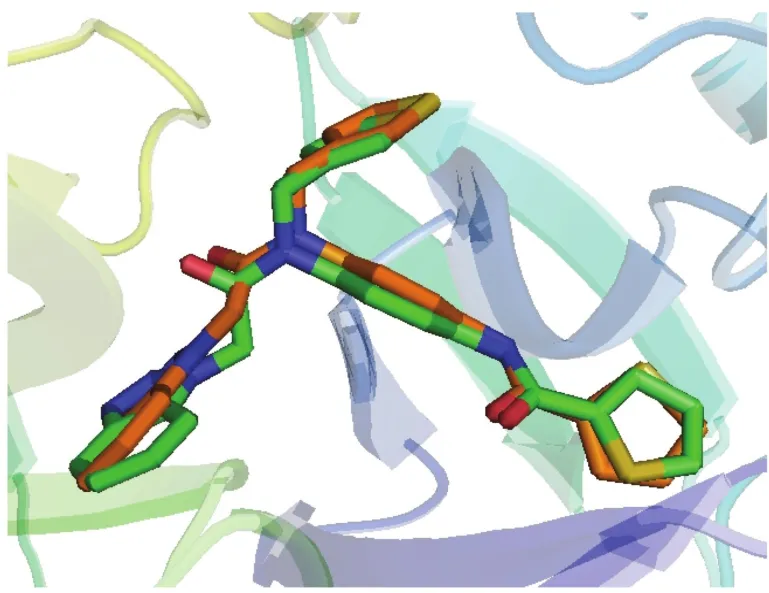
圖8 對接在3C-like蛋白酶中的1號分子的結合模式(橙色)和原1號分子(綠色)Fig.8 Binding mode of compound 1(orange)and the original molecule(green)docked in 3C-like protease

圖9 對接在3C-like蛋白酶中的抑制劑分子的結合模式Fig.9 Binding mode of inhibitor compound docked in 3C-like protease
與之前的CoMSIA等勢圖的結果結合分析,由CoMSIA氫鍵供體場的等勢圖(圖3(c))表明,在氫鍵供體場的積極作用域內,R2取代基上的氮原子作為氫鍵供體和結晶水HOH523形成氫鍵;而由CoMSIA氫鍵受體場的等勢圖(圖3(d))表明,在氫鍵受體場的積極作用域內,骨架上的氧原子和R3取代基上的靠近骨架部分的氮原子都作為氫鍵受體和氨基酸Glu169形成氫鍵。并且,CoMSIA靜電場的等勢圖(圖3(b))顯示,R4與R3取代基周圍分別存在有正電與負電云團,從對接結果中得知,在這兩個位置分別看到了正電性的氨基酸His41與Lys191和負電性的氨基酸Glu169。此外,由于Phe143、His166及His175等氨基酸中相對較大的苯環或雜環結構的存在,也使得在R3取代基附近引入大型取代基會產生較強的位阻效應,這與CoMSIA位阻場的等勢圖(圖3(a))中大塊的綠色云圖所代表的含義也是十分一致的。這些區域中氫鍵的形成以及正負電性與含苯環或雜環結構氨基酸的存在有利于提高生物的抑制活性。分子對接的結果與3D-QSAR等勢圖結果非常吻合,它們互為驗證和補充,充分說明了3D-QSAR模型的合理性。
同時,我們也將分子對接的結果與John等26已做的X射線晶體實驗結果(PDB ID:4YOI、4YOG與4YOJ)進行了對比。其中,氫鍵的對比結果如表2所示。從表中可以看出對接結果所得的四個氫鍵同樣在該實驗結果中出現,且它們的差別僅在作用力大小的不同,充分說明對接結果的正確性和穩定性。此外,他們實驗結果中的關鍵氨基酸,如氨基酸Met25、His41、Cys44、Ala46、Tyr54、Cys148、His166、Glu169和Gln192都出現在了分子對接的結合空腔中,也說明了分子對接是合理的,表明對接結果有很強的可靠性。
3.4分子動力學研究
對于配體分子結合機制的探明是一個重要的研究步驟,因此我們需要獲得肽類抑制劑小分子和3C-like蛋白酶大分子復合物更加真實的結合機制。分子動力學模擬正是能夠實現這種優化的方法之一51。分子動力學模擬可以有效地表達分子體系的狀態和行為隨時間的變化情況。相比屬于半柔性模擬方法的分子對接研究,分子動力學方法屬于動態模擬的過程,能夠高效迅速地搜索出配體分子的低能量構象,并對其構象變化的軌跡進行追蹤和表現。實驗中,我們采用了水環境下分子動力學模擬的方法來估計所得配體分子的結合親和性,并進一步評估分子對接中結合機制模型的可靠性。
3.4.11號分子的分子動力學研究
我們以分子對接中的配體-蛋白質復合物作為初始結構,對1號分子進行了50 ns的分子動力學研究,其發生構象改變的動力學圖像如圖10所示。其中,我們采取了通過對最初構象的監測從而對結構差異性進行幾何學測量的方法,均方根偏差(RMSD),來進一步地確保取樣方法的合理性與復合物大分子的動力學穩定性。圖10a分別展現了1號復合物(黑色)、蛋白(紅色)與1號配體(藍色)的分子動力學模擬的RMSD軌跡。圖中可以看出復合物與蛋白的RMSD軌跡極為接近,復合物的RMSD軌跡大部分被蛋白的RMSD軌跡覆蓋。它們的RMSD軌跡測量值范圍從0.010至0.050 nm,在25 ns時達到0.040 nm,且在此后的模擬過程中,該波動一直都保持在0.040 nm上下。而配體的RMSD軌跡,測量值范圍從0.005至0.030 nm,在5 ns后波動都穩定在0.020 nm左右。這些結果表明該分子動力學軌道具有良好的平衡性,并且系統中的分子對接復合物保持相對穩定。因此,我們采用1號復合物最后10 ns的平均結構來研究,相比于只采用單純的蛋白質晶體結構而言,會有更好的準確性和穩定性。同時,之前分子對接中得到的復合物也放在一起加以比較(圖10b),分子動力學和分子對接中得到的配體分子分別設為橙色和綠色的棍狀(圖10c)。從圖10b中我們可以直觀地看到,1號分子在分子動力學模擬與分子對接研究中占據的結合位點相同,且在構型構象上沒有明顯的差異,這印證了分子對接模型的合理性。但也有一點構象上的改變,仍不能忽視:在結合空腔中,分子動力學中配體的R1和R2取代基與分子對接相比,扭轉了一些角度。考慮到可能由于以上的構象變化而帶來的作用力的改變,我們對于分子動力學研究中得到復合物的結合機制也進行了相應分析。直觀的作用力構成如圖11所示,結合作用力主要包括疏水作用和四個氫鍵作用力。
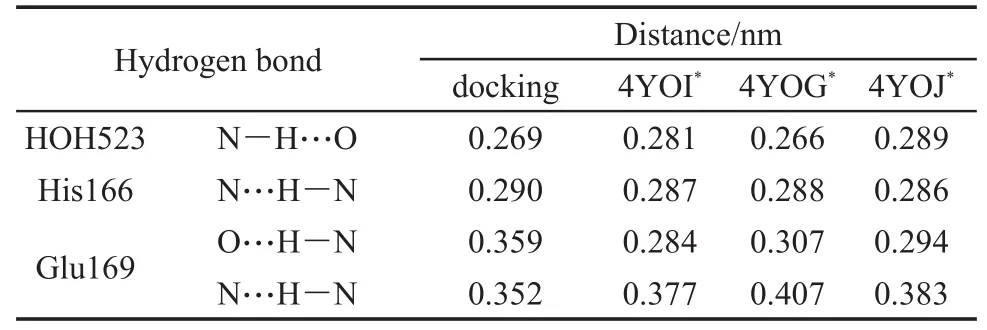
表2 分子對接和實驗結果的氫鍵距離Table 2 H-bond lengths from docking and experiment
從圖11中可以得出,1號分子的R2取代基部分固定在一個疏水的口袋之中,這個口袋主要由以下氨基酸殘基組成:Gly23、Ser24、Met25、Thr26、Leu49以及Ser147。而R3取代基部分也被一個疏水性的脂肪區域所包裹,組成這個區域的氨基酸主要包括 Phe143、Leu144、Cys145、Gly146、His166、Met168、Glu169以及Leu170。顯然,這兩個結合區域與分子對接中分析出的疏水位點S1和S2是相一致的。另一方面,分子動力學中配體分子結合的作用力與之前揭示的分子對接中的作用力是較為相似的,結晶水與氨基酸Glu169的重要作用力依然存在,說明了水分子介導的關鍵作用以及氨基酸Glu169的保守性,在一定程度上支持了分子對接過程中所得到的結論。但由于分子的R1和R2取代基扭轉了一些角度的原因使得分子與氨基酸His166的作用力消失,同樣與氨基酸Ser24產生了一個新的氫鍵作用力,使配體更有力地結合于對應位點中,而這并不影響分子整體的結合位點。具體分子對接與分子動力學氫鍵對比可由表3所示。總之,在水環境下的分子動力學研究中,1號分子在活性位點處保持穩定,所得的結果與分子對接結果也基本保持一致。

圖10 對接在3C-like蛋白酶中的1號分子的分子動力學模擬結果Fig.10 MD-simulated results of compound 1 docked in 3C-like protease
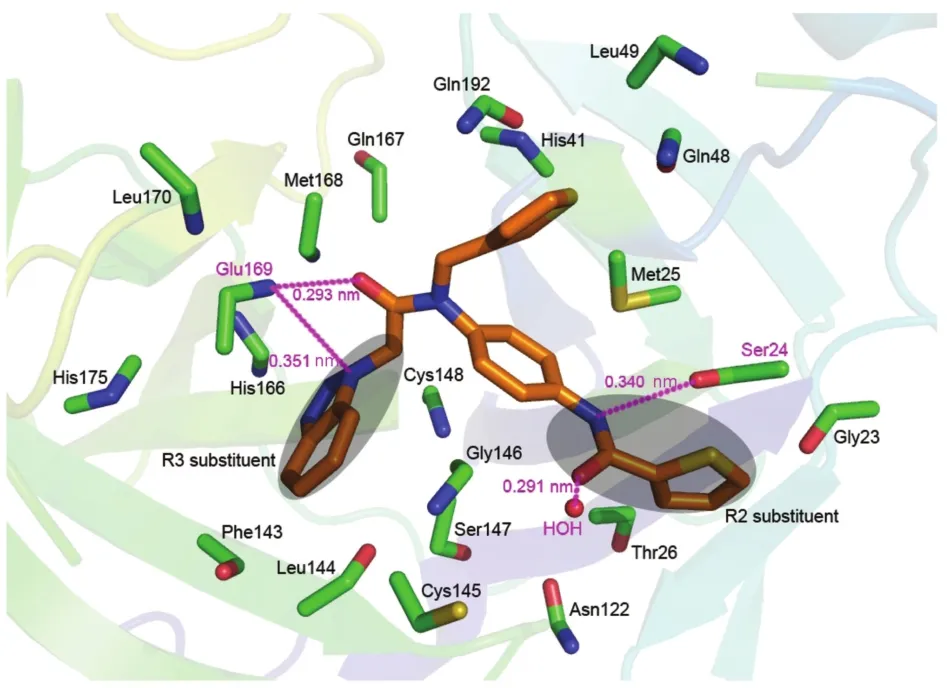
圖11 在水環境下分子動力學模擬的結合體的結合位點圖Fig.11 Plot of the in-water MD-simulated structures of the binding site

表3 分子對接與分子動力學模擬的氫鍵分析Table 3 H-bond analysis from docking and MD simulation
3.4.2 2號分子的分子動力學研究
除了1號分子的分子動力學研究,我們還對2號分子進行了50 ns分子動力學研究。2號復合物(黑色)、蛋白(紅色)與2號配體(藍色)的RMSD圖以及結合模式圖如圖12所示。在圖12a所示的RMSD圖譜中,復合物與蛋白的RMSD數值在10 ns后穩定在0.040 nm左右,而配體的數值則在20 ns后保持在0.020 nm上下,這顯示了一個穩定的分子動力學軌道。2號復合物最后10 ns的結合模式圖如圖12b所示,2號分子的R2取代基部分同樣固定在一個由Ser24、Met25、Thr26、Ala46、Asp47以及Leu49等氨基酸殘基組成的疏水口袋之中。而R3取代基部分也被一個由 Cys145、Gly146、Cys148、Met168、Glu169、Leu170以及His194等氨基酸組成的疏水脂肪區域所包裹。很顯然,這兩個小的疏水空腔與前文的位點S1和S2是基本相同的。這表明2號分子仍處于上述提及的疏水結合位點中。
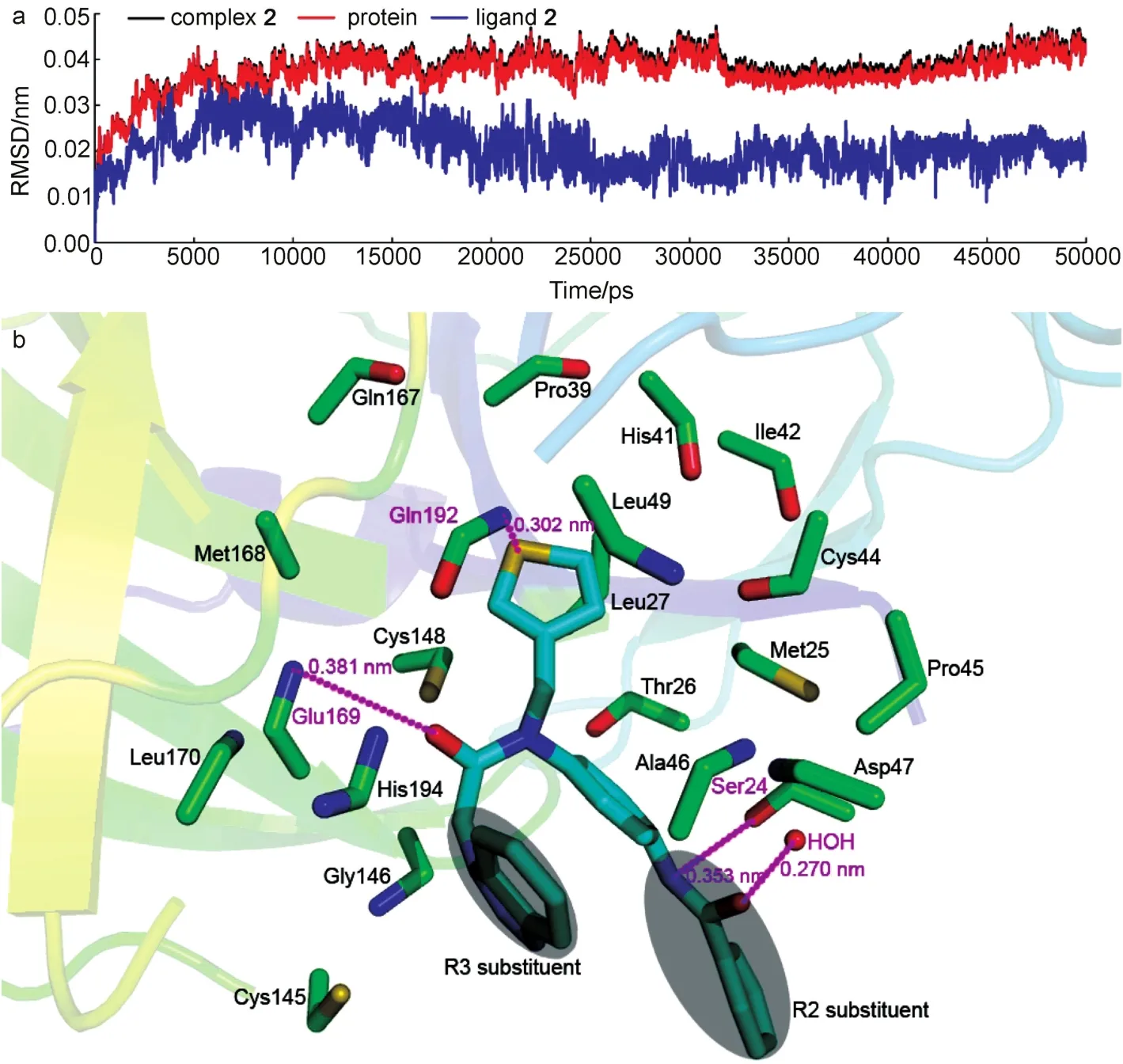
圖12 對接在3C-like蛋白酶中的2號分子的分子動力學模擬結果Fig.12 MD-simulated results of compound 2 docked in 3C-like protease
除了疏水作用,2號分子的分子動力學所得到的氫鍵作用列于表4中,其中也列出了1號分子的分子動力學中的氫鍵作用。對比表4的數據可以發現,它們各自四個氫鍵作用中有三個氫鍵相同,并且可以發現2號分子與Gln192殘基又有一個新的氫鍵作用。分子在結構方面的特征最終決定了它們各自動力學模擬時的特殊構造要求52。我們通過對比1號與2號分子結構之間的差異進行分析,它們的結構特征圖如圖13所示。由圖13可以看出,它們的R1、R3與R4取代基以及R2取代基靠近分子骨架部分擁有共同的結構,而它們的差別僅于R2取代基遠位部分,1號分子被噻吩環取代,而2號分子被苯環取代。大部分共同的結構使它們處于位阻、靜電、氫鍵供體與氫鍵受體場的積極區域有諸多相同,但是由于噻吩環中具有一個S原子可作為氫鍵受體益于分子活性,導致1號分子活性略高于2號分子。因此,以上1號與2號分子結構之間的微小差異導致了結合模式中氫鍵的細微改變,體現了R2取代基遠位部分在配體與蛋白結合過程中的重要性。綜上所述,在1號與2號分子的分子動力學研究中,配體分子在活性位點都保持穩定,所得的兩個結果也基本保持一致。
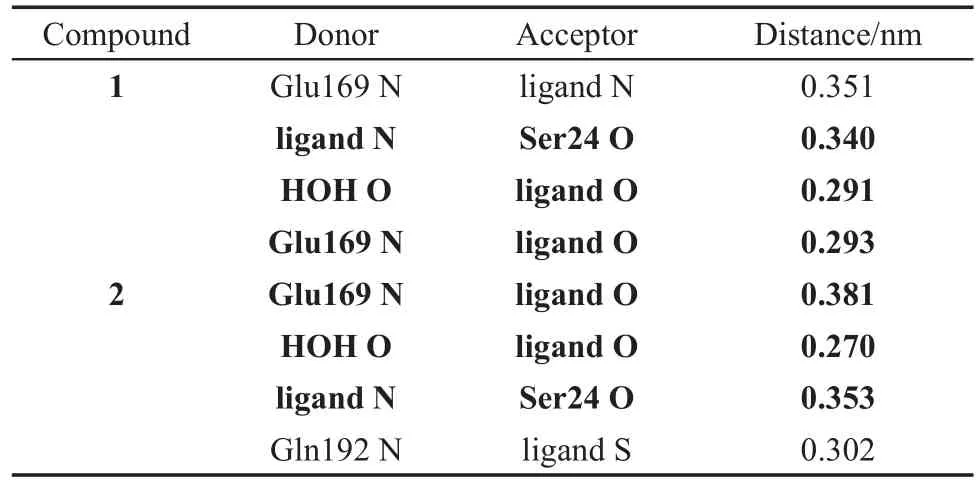
表4 1號與2號分子的分子動力學模擬的氫鍵分析Table 4 H-bond analysis from MD simulation of compound 1 and compound 2
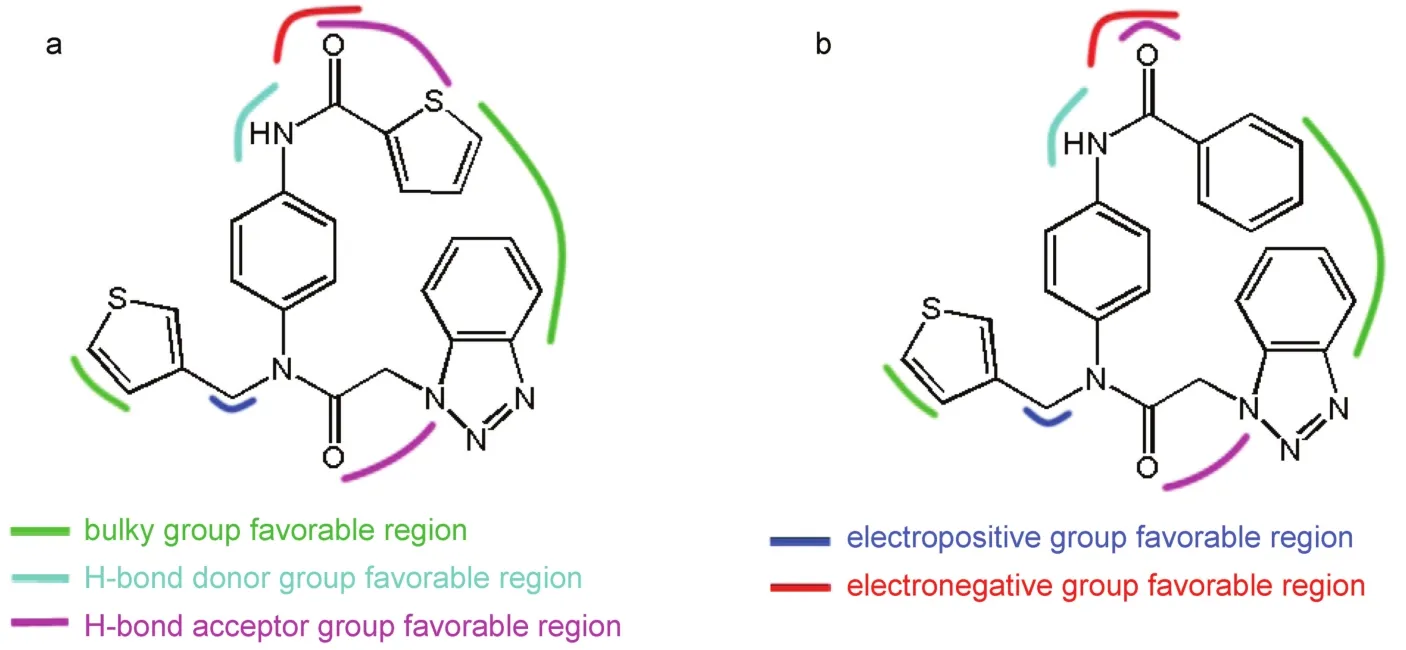
圖13 1號與2號分子的結構特征圖Fig.13 Structural features of compound 1 and compound 2
3.5新肽類化合物的設計與活性預測
基于43個肽類抑制劑對受體3C-like蛋白酶的抑制活性,建立了具有較強可靠性和良好預測能力的3D-QSAR模型。通過對模型的分析揭示了這類抑制劑分子結構上的特點,為更好地理解配體抑制劑和受體蛋白的作用機理提供了有效的幫助,同時為進一步優化此類抑制劑提供了可靠的依據。圖14顯示的是我們借助上述研究所獲得的基于3C-like蛋白酶上影響肽類分子抑制作用的關鍵性結構特征。該圖將有助于指導篩選和開發更優良的抗MERS-CoV藥物。
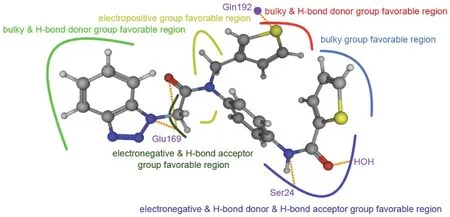
圖14 基于3C-like蛋白酶受體上肽類配體分子的交互特性Fig.14 Interaction features of peptidomimetic ligand molecule with the 3C-like protease receptor
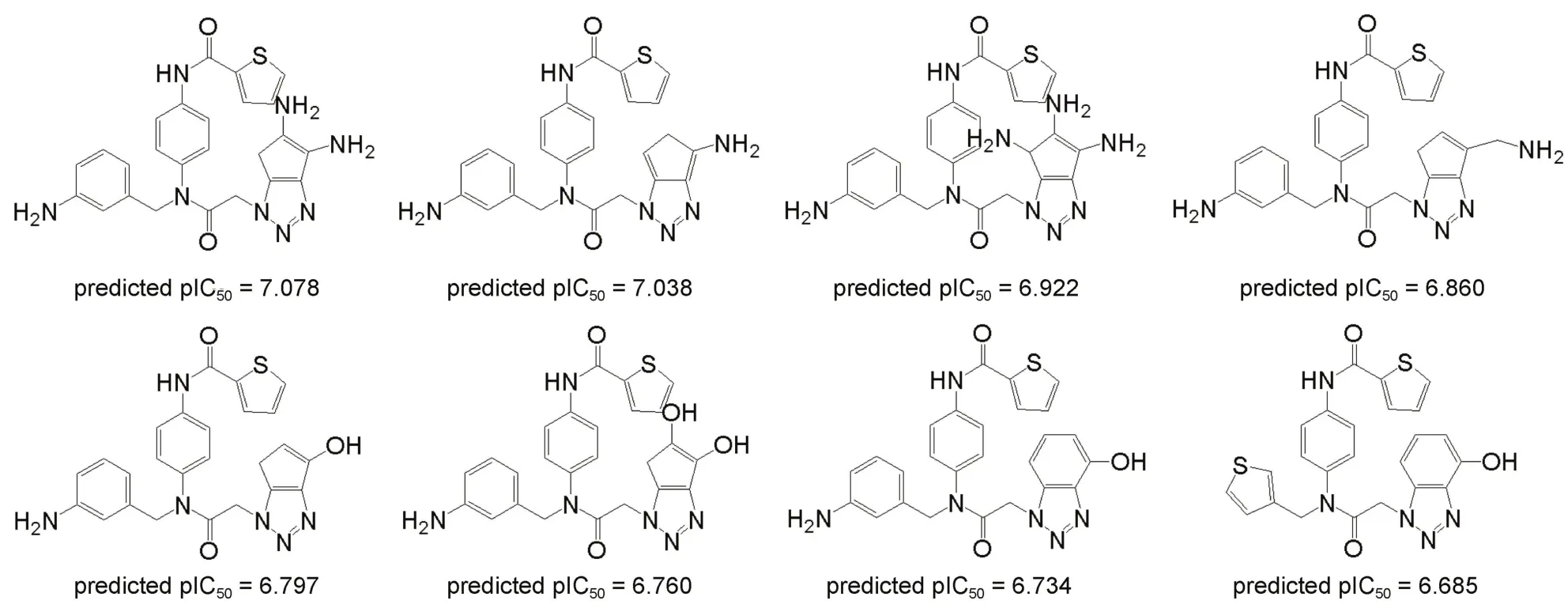
圖15 新設計分子的結構Fig.15 Structures of newly designed molecules
例如,以1號分子為模板,我們根據以上肽類分子的交互特性圖,通過修飾其各個取代基,設計了8個新的肽類3C-like蛋白酶抑制劑。這8個分子的分子結構如圖15所示,CoMSIA模型預測它們的pIC50值均高于模板分子(6.48),關于其具體的藥物活性還需通過實驗進一步驗證。
4 結論
以計算機輔助藥物設計方法的理論和手段,借助3D-QSAR建模、分子對接和分子動力學方法對一系列新合成的肽類3C-like蛋白酶抑制劑進行了分子構效關系以及配受體結合作用的研究。本文具體研究結論如下:
(1)建立了基于配體疊合的3D-QSAR模型,模型表現出較好的內部一致性,并得到了CoMSIA模型等統計指標;
(2)對于R1取代基,遠離分子骨架的部分(靠近R2取代基的周圍),大位阻基團和氫鍵供體基團有利于分子活性的提高;
(3)對于R2取代基,臨近分子骨架的部分,負電性基團、氫鍵供體基團和氫鍵受體基團有利于分子活性的提高;同樣,靠近R1取代基的周圍,大位阻基團也有利于分子活性的提高;
(4)對于R3取代基,臨近分子骨架的部分,負電性基團和氫鍵受體基團有利于分子活性的提高;同樣,遠離分子骨架的部分,大位阻基團和氫鍵供體基團也有利于分子活性的提高;
(5)對于R4取代基,臨近分子骨架的部分,正電性取代基有利于分子活性的提高;
(6)分子對接結果顯示,抑制劑分子通過疏水作用以及與結晶水HOH523、氨基酸His166和Glu169產生的四個氫鍵作用使小分子穩定在受體的空腔中,說明疏水性以及結晶水、氨基酸His166和Glu169在配體和受體結合過程中產生重要的作用;
(7)分子動力學結果不僅與分子對接結果有較一致的結合空腔,證明了分子對接結果的可靠性,而且在結合空腔中,發現了兩個新的關鍵氨基酸Ser24與Gln192,它們與配體產生了兩個較強的氫鍵。
結論表明我們的3D-QSAR、分子對接和分子動力學的研究得到了一個令人滿意的結果。此外,根據這些結論,一些新的具有潛在抑制活性的肽類化合物作為3C-like蛋白酶抑制劑被獲得。通過以上結論不但能夠深入了解肽類抑制劑的結構特征以及與3C-like蛋白酶的結合作用,也為今后它的實驗設計以及抗MERS-CoV藥物合成提供了新的方向。
Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Perlman,S.;Netland,J.Nat.Rev.Microbiol.2009,7,439. doi:10.1038/nrmicro2147
(2) Woo,P.C.;Huang,Y.;Lau,S.K.;Yuen,K.Y.Viruses 2010,2, 1804.doi:10.3390/v2081803
(3) Zaki,A.M.;Van Boheemen,S.;Bestebroer,T.M.;Osterhaus, A.D.;Fouchier,R.A.N.Engl.J.Med.2012,367,1814. doi:10.1056/NEJMoa1211721
(4) Rasmussen,S.A.;Gerber,S.I.;Swerdlow,D.L.Clin.Infect. Dis.2015,60,1686.doi:10.1093/cid/civ118
(5) Lau,S.K.;Li,K.S.;Tsang,A.K.;Lam,C.S.;Ahmed,S.; Chen,H.;Chan,K.H.;Woo,P.C.;Yuen,K.Y.J.Virol.2013, 87,8638.doi:10.1128/JVI.01055-13
(6) Chan,J.F.W.;Lau,S.K.P.;Woo,P.C.Y.J.Formos.Med. Assoc.2013,112,372.doi:10.1016/j.jfma.2013.05.010
(7) de Groot,R.J.;Baker,S.C.;Baric,R.S.;Brown,C.S.; Drosten,C.;Enjuanes,L.;Fouchier,R.A.;Galiano,M.; Gorbalenya,A.E.;Memish,Z.A.J.Virol.2013,87,7790. doi:10.1128/JVI.01244-13
(8) Haagmans,B.L.;Al Dhahiry,S.H.;Reusken,C.B.;Raj,V.S.; Galiano,M.;Myers,R.;Godeke,G.J.;Jonges,M.;Farag,E.; Diab,A.Lancet.Infect.Dis.2014,14,140.doi:10.1016/S1473-3099(13)70690-X
(9) Niemeyer,D.;Zillinger,T.;Muth,D.;Zielecki,F.;Horvath,G.; Suliman,T.;Barchet,W.;Weber,F.;Drosten,C.;Müller,M.A. J.Virol.2013,87,12489.doi:10.1128/JVI.01845-13
(10) Matthews,K.L.;Coleman,C.M.;van der Meer,Y.;Snijder,E. J.;Frieman,M.B.J.Gen.Virol.2014,95,874.doi:10.1099/ vir.0.062059-0
(11) Wang,Q.;Qi,J.;Yuan,Y.;Xuan,Y.;Han,P.;Wan,Y.;Ji,W.;Li, Y.;Wu,Y.;Wang,J.Cell Host Microbe 2014,16,328. doi:10.1016/j.chom.2014.08.009
(12) Woo,P.C.;Lau,S.K.;Li,K.S.;Tsang,A.K.;Yuen,K.Y. Emerg.Microbes.Infect.2012,1,e35.doi:10.1038/emi.2012.45
(13) Ziebuhr,J.;Snijder,E.J.;Gorbalenya,A.E.J.Gen.Virol.2000, 81,853.doi:10.1099/0022-1317-81-4-853
(14) Thiel,V.;Ivanov,K.A.;Putics,A.;Hertzig,T.;Schelle,B.; Bayer,S.;Wei?brich,B.;Snijder,E.J.;Rabenau,H.;Doerr,H. W.J.Gen.Virol.2003,84,2305.doi:10.1099/vir.0.19424-0
(15) Liu,W.;Zhu,H.M.;Niu,G.J.;Shi,E.Z.;Chen,J.;Sun,B.; Chen,W.Q.;Zhou,H.G.;Yang,C.Bioorg.Med.Chem.2014, 22,292.doi:10.1016/j.bmc.2013.11.028
(16) Kuo,C.J.;Liang,P.H.ChemBioEng Rev.2015,2,118. doi:10.1002/cben.201400031
(17) Chen,S.;Chen,L.;Tan,J.;Chen,J.;Du,L.;Sun,T.;Shen,J.; Chen,K.;Jiang,H.;Shen,X.J.Biol.Chem.2005,280,164. doi:10.1074/jbc.M408211200
(18) Ramajayam,R.;Tan,K.P.;Liu,H.G.;Liang,P.H.Bioorg. Med.Chem.Lett.2010,20,3569.doi:10.1016/j. bmcl.2010.04.118
(19) Thanigaimalai,P.;Konno,S.;Yamamoto,T.;Koiwai,Y.; Taguchi,A.;Takayama,K.;Yakushiji,F.;Akaji,K.;Chen,S.E.; Naser-Tavakolian,A.Eur.J.Med.Chem.2013,68,372. doi:10.1016/j.ejmech.2013.07.037
(20) Kang,C.M.;Zhao,X.H.;Wang,X.Y.;Cheng,J.G.;Lü,Y.T. Acta Phys.-Chim.Sin.2013,29,431.[康從民,趙緒浩,王新宇,程家高,呂英濤.物理化學學報,2013,29,431.]doi:10.3866/ PKU.WHXB201211151
(21) Zhang,S.Z.;Zheng,C.;Zhu,C.J.Acta Phys.-Chim.Sin.2015, 31,2395.[張淑貞,鄭 超,朱長進.物理化學學報,2015,31, 2395.]doi:10.3866/PKU.WHXB201510142
(22) Pillaiyar,T.;Manickam,M.;Jung,S.H.Med.Chem.2015,5, 361.doi:10.4172/2161-0444.1000287
(23) Ren,Z.;Yan,L.;Zhang,N.;Guo,Y.;Yang,C.;Lou,Z.;Rao,Z. Protein Cell 2013,4,248.doi:10.1007/s13238-013-2841-3
(24) Deng,X.;StJohn,S.E.;Osswald,H.L.;O'Brien,A.;Banach,B. S.;Sleeman,K.;Ghosh,A.K.;Mesecar,A.D.;Baker,S.C.J. Virol.2014,88,11886.doi:10.1128/JVI.01528-14
(25) Tomar,S.;Johnston,M.L.;John,S.E.S.;Osswald,H.L.; Nyalapatla,P.R.;Paul,L.N.;Ghosh,A.K.;Denison,M.R.; Mesecar,A.D.J.Biol.Chem.2015,290,19403.doi:10.1074/ jbc.M115.651463
(26) John,S.E.S.;Tomar,S.;Stauffer,S.R.;Mesecar,A.D.Bioorg. Med.Chem.2015,23,6036.doi:10.1016/j.bmc.2015.06.039
(27) AbdulHameed,M.D.M.;Hamza,A.;Liu,J.J.;Zhan,C.G.J. Chem.Inf.Model.2008,48,1760.doi:10.1021/ci800147v
(28) Gasteiger,J.;Marsili,M.Tetrahedron 1980,36,3219. doi:10.1016/0040-4020(80)80168-2
(29) Clark,M.;Cramer,R.D.;Van Opdenbosch,N.J.Comput. Chem.1989,10,982.doi:10.1002/jcc.540100804
(30) Cramer,R.D.;Patterson,D.E.;Bunce,J.D.J.Am.Chem.Soc. 1988,110,5959.doi:10.1021/ja00226a005
(31) Klebe,G.;Abraham,U.;Mietzner,T.J.Med.Chem.1994,37, 4130.doi:10.1021/jm00050a010
(32) Edraki,N.;Das,U.;Hemateenejad,B.;Dimmock,J.R.Iran.J. Pharm.Res.2016,15,425.
(33) Li,X.L.;Ye,L.;Wang,X.X.;Wang,X.Z.;Liu,H.L.;Qian,X. P.;Zhu,Y.L.;Yu,H.X.Sci.Total Environ.2012,441,230. doi:10.1016/j.scitotenv.2012.08.072
(34) Shah,P.;Saquib,M.;Sharma,S.;Husain,I.;Sharma,S.K.; Singh,V.;Srivastava,R.;Shaw,A.K.;Siddiqi,M.I.Bioorg. Chem.2015,59,91.doi:10.1016/j.bioorg.2015.02.001
(35) Zhang,S.;Hou,B.;Yang,H.;Zuo,Z.Arch.Pharm.Res.2016, 1.doi:10.1007/s12272-016-0709-9
(36) Berman,H.M.;Battistuz,T.;Bhat,T.N.;Bluhm,W.F.;Bourne, P.E.;Burkhardt,K.;Iype,L.;Jain,S.;Fagan,P.;Marvin,J.; Padilla,D.;Ravichandran,V.;Schneider,B.;Thanki,N.; Weissig,H.;Westbrook,J.D.;Zardecki,C.Acta Crystallogr.D 2002,58,899.doi:10.1107/S0907444902003451
(37) Verdonk,M.L.;Cole,J.C.;Hartshorn,M.J.;Murray,C.W.; Taylor,R.D.Proteins 2003,52,609.doi:10.1002/prot.10465
(38) Duan,A.X.;Chen,J.;Liu,H.D.;Liu,X.H.;Lu,X.Q.J.Mol. Sci.2009,25,473.[段愛霞,陳 晶,劉宏德,劉秀輝,盧小泉.分析科學學報,2009,25,473.]
(39) Arooj,M.;Sakkiah,S.;Kim,S.;Arulalapperumal,V.;Lee,K. W.PloS One 2013,8,e63030.doi:10.1371/journal. pone.0063030
(40) Van der Spoel,D.;Lindahl,E.;Hess,B.;Groenhof,G.;Mark,A. E.;Berendsen,H.J.C.J.Comput.Chem.2005,26,1701. doi:10.1002/jcc.20291
(41) Lindahl,E.;Hess,B.;Van Der Spoel,D.J.Mol.Model.2001,7, 306.doi:10.1007/s008940100045
(42) vanAalten,D.M.F.;Bywater,R.;Findlay,J.B.C.;Hendlich, M.;Hooft,R.W.W.;Vriend,G.J.Comput.Aid.Mol.Des. 1996,10,255.doi:10.1007/BF00355047
(43) Berendsen,H.J.C.;Postma,J.P.M.;van Gunsteren,W.F.; Hermans,J.Interaction Models for Water in Relation to Protein Hydration.In Intermolecular Forces;Springer Netherlands: Berlin,1981;pp 331-342.
(44) Ryckaert,J.P.;Ciccotti,G.;Berendsen,H.J.C.J.Comput. Phys.1977,23,327.doi:10.1016/0021-9991(77)90098-5
(45) Essmann,U.;Perera,L.;Berkowitz,M.L.;Darden,T.;Lee,H.; Pedersen,L.G.J.Chem.Phys.1995,103,8577.doi:10.1063/ 1.470117
(46) Alexander,G.;Alexander,T.J.Mol.Graph.Model.2002,20, 269.doi:10.1016/S1093-3263(01)00123-1
(47) Kamsri,P.;Punkvang,A.;Hannongbua,S.;Saparpakorn,P.; Pungpo,P.RSC Adv.2015,5,52926.doi:10.1039/C5RA08103C
(48) Gao,X.;Han,L.;Ren,Y.Molecules 2016,21,591.doi:10.3390/ molecules21050591
(49) Da,C.X.;Mooberry,S.L.;Gupton,J.T.;Kellogg,G.E. J.Med.Chem.2013,56,7382.doi:10.1021/jm400954h
(50) Jeffrey,G.A.An Introduction to Hydrogen Bonding.In Topics in Physical Chemistry;Oxford University Press:NewYork,1997.
(51) Minini,L.;Alvarez,G.;González,M.;Cerecetto,H.;Merlino, A.J.Mol.Graph.Model.2015,58,40.doi:10.1016/j. jmgm.2015.02.002
(52) Wang,J.;Li,F.;Li,Y.;Yang,Y.;Zhang,S.;Yang,L.Mol. BioSyst.2013,9,2296.doi:10.1039/c3mb70105k
QSAR,Molecular Docking and Molecular Dynamics of 3C-like Protease Inhibitors
LIN Feng1FU Xin-Mei2,*WANG Chao1JIANG Si-Yu1WANG Jing-Hui1ZHANG Shu-Wei1YANG Ling3LIYan1,*
(1School of Chemical Engineering,Dalian University of Technology,Dalian 116024,Liaoning Province,P.R.China;2State Key Laboratory of Fine Chemicals,Dalian University of Technology,Dalian 116024,Liaoning Province,P.R.China;3Laboratory of Pharmaceutical Resource Discovery,Dalian Institute of Chemical Physics,Chinese Academy of Sciences, Dalian 116023,Liaoning Province,P.R.China)
3C-like protease is an extremely important protease involved in the multiplicative process of coronaviruses,includingthedeadlyMiddleEast respiratorysyndromecoronavirus(MERS-CoV).3C-likeprotease has become a hot research topic in the field of coronavirology.For the first time,a set of ligand-and receptorbased three-dimensional quantitative structure-activity relationships(3D-QSAR)models were carried out via comparative molecular field analysis(CoMFA)and comparative molecular similarity indices analysis(CoMSIA)to explore the structure-activity correlation of 43 peptidomimetic inhibitors of the 3C-like protease of the bat coronavirus HKU4(HKU4-CoV),which belongs to the same 2c lineage as MERS-CoV and shows high sequence similarity with MERS-CoV.Based on the ligand-based alignment,an optimal CoMSIAmodel(yielded by steric, electrostatic,H-bond donor and H-bond acceptor fields)was obtained with good predictive power of Q2=0.522, R2ncv=0.996 and R2pre=0.904(Q2:cross-validated correlation coefficient,R2ncv:non-cross-validated correlation coefficient,R2pre:predicted correlation coefficient for the test set of compounds).Molecular docking and molecular dynamics simulations were performed according to this model to further determine the interaction mechanism between ligands and the receptor.The experimental results show:(1)based on the optimal CoMSIAmodel,the 3D contour maps vividly illustrate that the molecular biological activity is influenced by the steric,electrostatic, H-bond donor and H-bond acceptor interactions of molecular groups.(2)Based on the docking analysis, hydrophobicity,crystal water,His166andGlu169haveimportantrolesintheligandsandreceptorbindingprocess. (3)Molecular dynamics(MD)simulations were carried out for further verification of the reliability of the docking model,and provide two new key residues,Ser24 and Gln192,which have two strong hydrogen bonds with the ligands.Some new compounds were obtained based on the modeling that are potential peptidomimetic inhibitors of 3C-like protease.These results help establish the binding mechanism between 3C-like protease and peptidomimetic inhibitors,and provide a valuable reference for future anti-MERS-CoV drug design.
MERS-CoV;3C-like protease;Peptidomimetic inhibitor;3D-QSAR;Molecular docking; Molecular dynamics
O641
10.3866/PKU.WHXB201608121
Received:May 31,2016;Revised:August 9,2016;Published online:August 12,2016.
*Corresponding authors.FU Xin-Mei,Email:fuxinmei@dlut.edu.cn;Tel:+86-411-84986206.LI Yan,Email:yanli@dlut.edu.cn; Tel:+86-411-84986062.
The project was supported by the National Natural Science Foundation of China(11201049).
國家自然科學基金(11201049)資助項目
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
