堿金屬修飾g-C3N4的能帶結構調控與載流子遷移過程
2016-11-22 09:48:52馬新國徐國旺黃楚云
物理化學學報 2016年10期
關鍵詞:結構
祝 林 馬新國,* 劉 娜 徐國旺 黃楚云,*
(1湖北工業大學理學院,武漢430068;2湖北工業大學太陽能高效湖北省協同創新中心,武漢430068)
堿金屬修飾g-C3N4的能帶結構調控與載流子遷移過程
祝林1,2馬新國1,2,*劉娜1徐國旺1,2黃楚云1,2,*
(1湖北工業大學理學院,武漢430068;2湖北工業大學太陽能高效湖北省協同創新中心,武漢430068)
采用平面波超軟贗勢方法研究了Li、Na、K三種堿金屬離子修飾對g-C3N4能帶結構和載流子遷移過程的影響。對建立的六種吸附構型分別采用廣義梯度近似和局域密度近似進行計算,發現三種堿金屬離子均更趨向于吸附在g-C3N4片層內的大空洞中央位置(F位置)。對于堿金屬與g-C3N4形成的n型Schottky結,通過能帶結構和功函數的計算,發現界面電荷的平衡使g-C3N4的能帶電位分別整體下移1.52 V(Li)、1.07 V (Na)、0.86 V(K)。其中K離子的引入一方面將g-C3N4的價帶和導帶調整到更合適的氧化還原電位,另一方面增大了g-C3N4的最高占據軌道(HOMO)和最低未占據軌道(LOMO)的分布,有利于提高載流子的遷移率,同時出現的HOMO和LOMO軌道非共面特性有利于電子和空穴的分離。
光催化;g-C3N4;能帶結構;載流子遷移
1 引言
尋找具有可以直接光催化裂解水和降解污染物的高活性半導體光催化劑是當前光催化領域研究的熱點之一。發生光催化反應的先決條件是具有強的可見光吸收和合適的氧化還原電位1,2,即要求光催化材料在電子特性上具有特殊的半導體特征。TiO2被認為是最有潛力的半導體光催化材料,但是過寬的帶隙以及量子產率不高等不易克服的缺點限制了其廣泛應用。多元金屬氧化物光催化材料一度也是光催化研究領域的熱點,如InMO4(M=Nb,Ta)3、BiVO44,5、Bi2WO66、Ga1-xZnxN1-xOx7、以及我們前期研究的BiPO48和Ag3PO49,但要實現其應用目標還很遠。最近發現部分層狀聚合物半導體是光催化候選材料,如類石墨層狀聚合物C3N4的帶隙為2.7 eV,其價帶頂和導帶底具有較合適的氧化還原電位,表現出一定的光催化性能10,11。同時,它還具有制備簡單、環境友好、適合規模化生產等優點12,13。
盡管如此,進一步提高g-C3N4的可見光吸收及載流子遷移率,以改善光電轉換效率仍然面臨著挑戰14。由于g-C3N4是一種類石墨的六方晶系層狀結構,且層內存在較大空洞,因此具有很高的比表面積和分散性,進行化學摻雜是一種有效調控g-C3N4電子結構和表面性能的有效手段。在過去的實驗研究中發現,共軛聚合物半導體在異質元素的修飾下,有可能窄化帶隙和降低HOMO軌道15,以及我們報道的S和P非金屬摻雜不僅可以提高可見光吸收,而且為載流子的輸運通道由原本的C―N―C改變為C―N―P―N―C16,克服了橋位N原子對載流子遷移限制作用,從而提高其遷移率。此外,還有報道I修飾g-C3N4增大了可見光吸收至600 nm,且增大了表面17;S修飾g-C3N4降低了過電位,提高了陽極光催化活性18;通過控制g-C3N4形變,提高了可見光吸收,且增強了載流子遷移19。
與過去的質子化摻雜和非金屬的共價摻雜不同,具有非局域化特性的金屬修飾g-C3N4,一定程度上可以活化g-C3N4表面,因此金屬修飾方法成為g-C3N4改性的新研究熱點。最近發現Fe離子修飾g-C3N4有更好的光催化活性,其作用機理為引入的Fe離子與N形成配位鍵,從而改變了g-C3N4的電子結構,降低了帶隙,提高了可見光吸收20,堿金屬K21和Na22修飾g-C3N4同樣也獲得較好的光催化活性,堿金屬的引入使能帶整體下移,提高了價帶的氧化能力,此外還促進了載流子的分離。然而堿金屬與周圍N原子的作用以及對能帶結構的調控機理仍缺乏研究,載流子的遷移過程仍需探討。因此,本文從g-C3N4的基本結構出發,采用平面波超軟贗勢方法計算了堿金屬在層狀聚合物g-C3N4內六種吸附構型的吸附能;并通過能帶結構和功函數計算,確定出堿金屬吸附對g-C3N4能帶結構及載流子遷移過程的影響。這些結果的獲得將為新型高效光催化體系的設計提供理論依據和新思路。
2 物理模型和計算方法
層狀聚合物g-C3N4是一種空間群為cmc21(No. 36)的六方晶體,由氮原子連接三均三嗪(C6N7)組成ABAB層疊結構,一個晶胞包含56個原子,其中N原子為32個,C原子為24個。所有的C原子與周圍的3個N原子形成三配位,橋位和中心位置的N原子與周圍的3個C原子形成三配位,而邊緣N原子與周圍的2個C原子形成二配位。由于堿金屬原子,尤其是K、Na原子的半徑較大(DLi=0.15 nm,DNa=0.19 nm,DK=0.23 nm),而C3N4環的直徑僅為0.22 nm,因此,若摻入的堿金屬在g-C3N4片層內,則僅有可能在N圍成的大空洞中。若摻入的堿金屬在片層內,最有代表性的位置是在原子的正上方。因此,我們選取了位于A、B、C、D、E原子的正上方的5種吸附構型;以及位于片層內N圍成的大空洞中心的構型,共確定了6種可能的吸附構型,所有的吸附位置如圖1所示。
采用基于密度泛函理論的平面波超軟贗勢方法研究結構的穩定性及電學性質23。交換關聯能采用廣義梯度近似(GGA)24。為了能正確描述層間的范德華力,選用一種雜化的半經驗方法(OBS)處理衰減原子對的離散修正量C6R-6,該修正方案可以較好平衡在處理該離散項上計算時間的花費與改善非成鍵作用之間的需要25。原子-電子間的相互作用采用了Vanderbilt方案的超軟贗勢26。在描述原子實與價電子之間的相互作用時,選取的價電子組態分別為C:2s22p2,N:2s22p3,Li:1s22s1,Na:2s22p63s1,K:3s23p64s1,其他軌道電子視為芯電子進行計算。平面波截斷能設置為340 eV,第一布里淵區采用Monkhorst的5×5×4。結構優化選用的自恰收斂精度設置為1×10-6eV·atom-1,原子受力不超過0.01 eV,最大原子位移不超過5× 10-5nm。
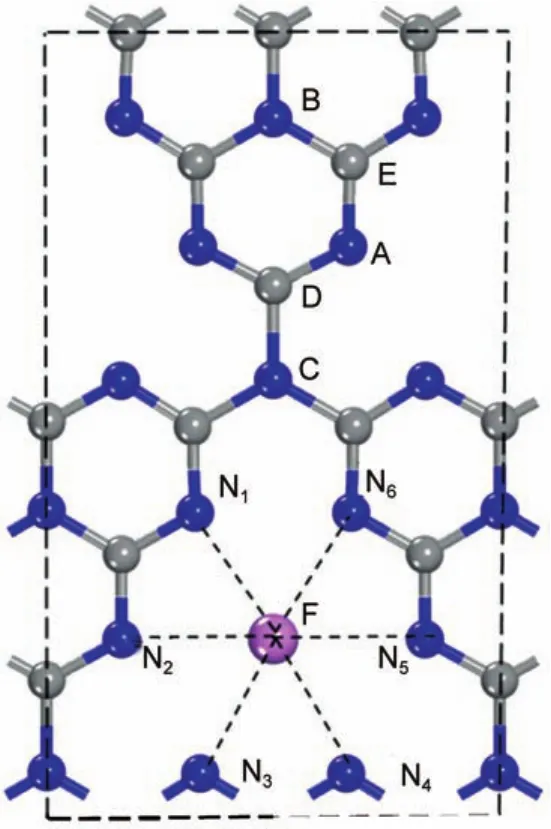
圖1 堿金屬吸附于g-C3N4的六種摻雜構型俯視圖Fig.1 Top views of the six doped configurations of alkali metal adsorbed on g-C3N4The lettersA-F represent the absorption positions of alkali metal atom,respectively.
3 結果和討論
3.1結構的穩定性
運用LDA和GGA方法優化后,結果顯示g-C3N4具有穩定的平面層狀結構,這與單層g-C3N4容易形成弓形的幾何結構完全不同27,23。與實驗結果相比較,眾所周知采用LDA方法往往會低估晶體結構常數,高估體系的結合能,而GGA方法剛好相反。前者與文獻28,29中的理論結果比較一致。Gracia和Kroll29采用分子動力學方法優化的結果顯示在面內相臨的三均三嗪中心N原子之間的距離為0.677 nm,這個結果與Wang等10采用XRD方法獲得結果(0.681 nm)比較一致。這兩個結果與Bojdys等12的實驗結果(0.730 nm)有很大出入。認為實際體相的g-C3N4中,晶體結構參數應該在LDA和GGA兩種方法計算的結果之間,因此可以估算面內相臨的三均三嗪中心N原子之間的距離和層間距離分別為0.714和0.319 nm。
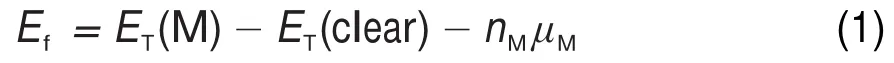
為獲得結構合理且穩定的吸附構型,對建立的6種摻雜構型進行了吸附能的計算。堿金屬在g-C3N4表面上的吸附能定義為其中ET(M)為M原子吸附在化學計量比結構內的體系總能量,ET(clear)為清潔化學計量比結構的體系總能量,nM為吸附的堿金屬原子數,μM為吸附的堿金屬原子的絕對化學勢。當吸附能為負數時,表示該吸附為放熱過程,且數值越大表明吸附作用越強,吸附結構越穩定。
根據(1)式,計算出了Li、Na、K三種堿金屬6種不同摻雜構型的吸附能,列于表1中。可以看出,對于Li、Na、K離子摻雜所選取的A-F共6種不同摻雜構型中,構型F的吸附能比其他的AE吸附構型分別要高1.8(Li)、3.6(Na)和5.3 eV(K)以上。吸附構型F的吸附能均為負數,且絕對值最大,即最大的吸附能分別為Ef(Li)=-4.477 eV,Ef(Na)=-4.144 eV,Ef(K)=-3.603 eV。這些結果均說明了吸附構型F的結構最為穩定,并且摻雜易于實現。為了更精確地確定吸附構型F中堿金屬原子所處的位置,將結構優化后的堿金屬原子M到周圍最鄰近6個N原子的距離列于表2中。結果顯示,M到周圍N原子的距離表現出高度的對稱性,即M―N1和M―N2距離相等,M―N3和M―N6距離相等,M―N4和M―N5距離相等,這說明堿金屬M完全處于N六邊形的空洞中心,而且位于平面內。這個結果與Hu等30基于實驗的推測是一致的。
3.2Schottky結能級調制
為了探討摻雜構型F會對g-C3N4的能帶結構帶來的影響,分別采用LDA和GGA方法計算了純g-C3N4的能帶結構和摻雜后的能帶結構,所得帶隙值記錄在表3中。從結果上看,采用LDA和GGA方法所得的帶隙值均比實驗值要小,而與Xiong31和Aspera32等的理論結果比較一致,這是由于DFT在計算帶隙值時普遍存在不可避免的誤差。堿金屬的引入對能帶的形狀影響不大,堿金屬摻入前后,能帶結構的骨架基本未變,而帶隙有明顯減小,這說明吸收譜的范圍會有一定的紅移,這與Hu等30的實驗結果和Xiong等31的理論結果相符;同時可以發現,隨著堿金屬原子半徑的增大,對帶隙的窄化能力逐步減弱。此外,這種窄化應該是與摻入堿金屬的濃度有關的,在Hu等30的實驗結果中,K摻雜g-C3N4的帶隙值隨著K濃度的變化而在2.52-2.65 eV間變化。
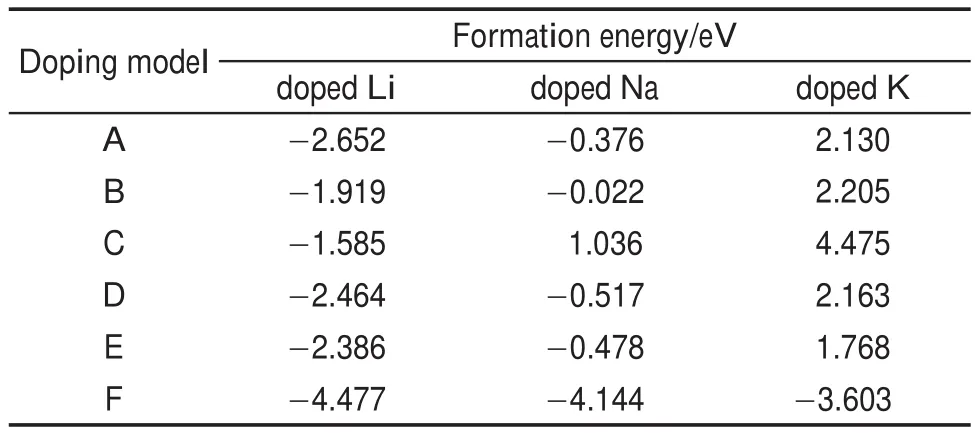
表1 三種堿金屬分別摻雜g-C3N4時,6種不同吸附構型(A-F)的吸附能Table 1 Adsorption energies of six different adsorption configurations(A-F)for doped g-C3N4with three alkali metals

表2 吸附構型F中堿金屬離子M與周圍6個N原子的距離及堿金屬M和6個N原子的Milliken布居數Table 2 Distances between the alkali metal ion M and its neighboring six N atoms and the Milliken population of alkali metal ion M and its neighboring six N atoms for adsorption configuration F
空洞周圍N原子的2p軌道對價帶頂和導帶底都有貢獻。因此,堿金屬引入導致這些N原子的電荷分布發生改變,勢必對g-C3N4原有費米能級的位置產生一定的影響。一般而言,金屬和半導體表面接觸形成Schottky結,在界面上形成表面勢壘。由于半導體的費米能級較高,半導體的費米能級隨著整個能帶結構移動到與金屬的費米能級相平。為了定量分析Schottky結的能級調制,計算了純g-C3N4以及堿金屬摻雜后g-C3N4的功函數。其物理意義是把一個電子從固體內部移到此物體表面所需的最少的能量,其值也等于費米能級對絕對真空能級(AVS)的電位,在這里用于確定摻雜前后g-C3N4費米能級的位置。
針對前面的物理模型,建立(001)晶面的Slab結構(真空為1.5 nm),獲得的純g-C3N4功函數為4.42 eV,Li、Na、K摻雜g-C3N4的功函數為5.94、5.57和5.32 eV,其結果如圖2所示。由于氫還原能級(NHE)與絕對真空能級(AVS)間的相對因子為4.5 eV,換算后費米能級的電位分別為-0.08、1.44、1.07和0.82 V(vs NHE)。可以看出摻雜后,g-C3N4費米能級在向正向移動。同時,隨著堿金屬原子半徑的增大,費米能級下降的幅度逐次減小。
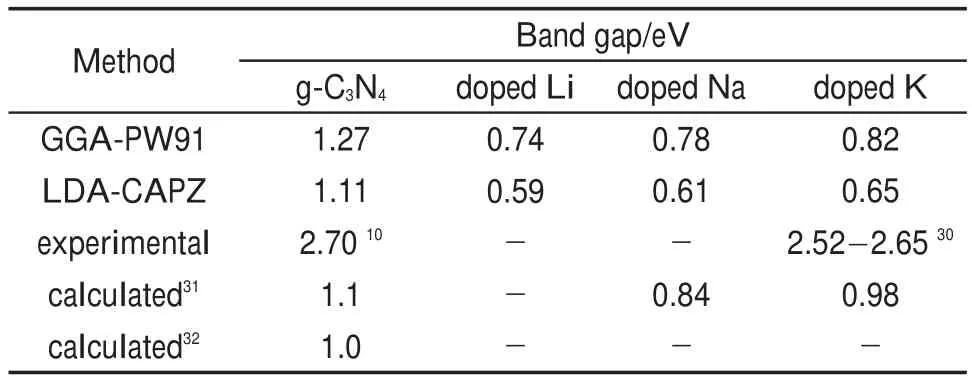
表3 摻雜前后g-C3N4的帶隙值Table 3 Band gap of g-C3N4before and after doping
導帶邊電位采用電化學方法測量是一件比較困難的事情。為了直接從理論上獲得導帶邊和價帶邊的電位(真空電位),運用原子平均電負性方法以及實驗帶隙值通過下列式子進行估算33,34
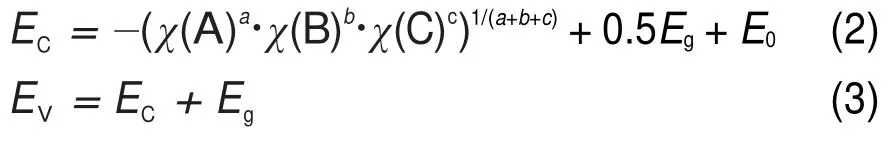
這里χ(A)、χ(B)和χ(C)分別為原子A、B和C的絕對電負性,EC、EV和Eg分別為導帶底電位、價帶頂電位和帶隙值,這里真空電位與氫還原電位差E0為4.44 V。一般而言,在溫度T=298 K,氣壓為1.013×105Pa時,許多半導體(如Ta3N5,GaN等)在溶劑中與H+或者OH-離子反應,結果界面上的帶邊將隨溶劑的pH值按照大約60 mV·pH-1的線性關系而變化33,35,這主要是由于表面吸附的H+或者OH-
離子的驅動作用。在pH=pHZPC,計算獲得純g-C3N4的導帶邊和價帶邊電位(相對氫還原)分別為-1.05和1.65 V。
當向g-C3N4中摻入堿金屬以后,g-C3N4的費米能級將下移,堿金屬的費米能級將上移,直至兩者費米能級拉平,由于Schottky結作用,g-C3N4的能帶整體下移,即價帶頂電位向正向移動,其光生空穴的氧化性將增加,同時導帶底電位也向正向移動,其光生電子的還原能力有所減弱。其中Li、Na的摻雜使g-C3N4的導帶電位降低到0 V以下,過大的能級調制使其還原能力有所不足,而K摻雜對g-C3N4導帶底的電位降低至-0.09 V,而價帶頂的電位調整為2.41 V,可以滿足光催化反應過程中光生電子和空穴的電位要求1,2。事實上,這種電位的調制應該與摻入堿金屬離子的濃度有相當大的關系,Hu等30通過控制K離子的濃度使g-C3N4的導帶和價帶電位在-1.09-1.56 V和-0.31-2.21 V間變化。為了清楚地了解在同一濃度下,三種堿金屬引入所引起的能級變化,將純g-C3N4和堿金屬摻雜后g-C3N4的價帶和導帶電位總結于圖3中。
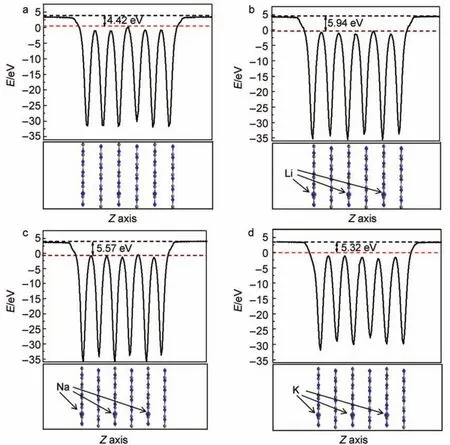
圖2 沿著垂直g-C3N4(001)晶面方向的功函數Fig.2 Work functions in the vertical direction of g-C3N4(001)plane (a)pure g-C3N4;(b)Li doped g-C3N4;(c)Na doped g-C3N4;(d)K doped g-C3N4
3.3載流子遷移過程
堿金屬摻雜對g-C3N4的能帶結構顯示出一定的調制作用,這必然對光生載流子的遷移過程的產生重要影響。首先分析了純g-C3N4和堿金屬摻雜g-C3N4的分波態密度(PDOS),如圖4所示。可以看出,純g-C3N4和堿金屬摻雜g-C3N4的導帶底附近均是由N 2p和C 2p軌道雜化構成,而價帶頂附近均來自N 2p態的貢獻,少量來自C的貢獻。尤其值得注意的是,堿金屬的雜質能級位置很深,在價帶頂和導帶底附近均無態密度分布,因此可以預見堿金屬離子并不直接參加光生載流子的產生、分離和轉移過程。那么,堿金屬的引入對g-C3N4電子結構的影響應該來自于堿金屬對周圍鄰近N原子的電荷重新分布。為了證明這種觀點,對鄰近N原子的Mulliken原子布居定量地進行了分析,如表2所示。可以看出,隨著由Li到K堿金屬原子的金屬性增強,Mulliken布居數依次增大,且數值都大于1,這說明堿金屬不僅失去了外層電子,次外層電子也失去了一部分;而且這些失去的電子大部分轉移給了周圍的N原子。顯然,堿金屬的引入對周圍N原子的電荷分布有重要影響。
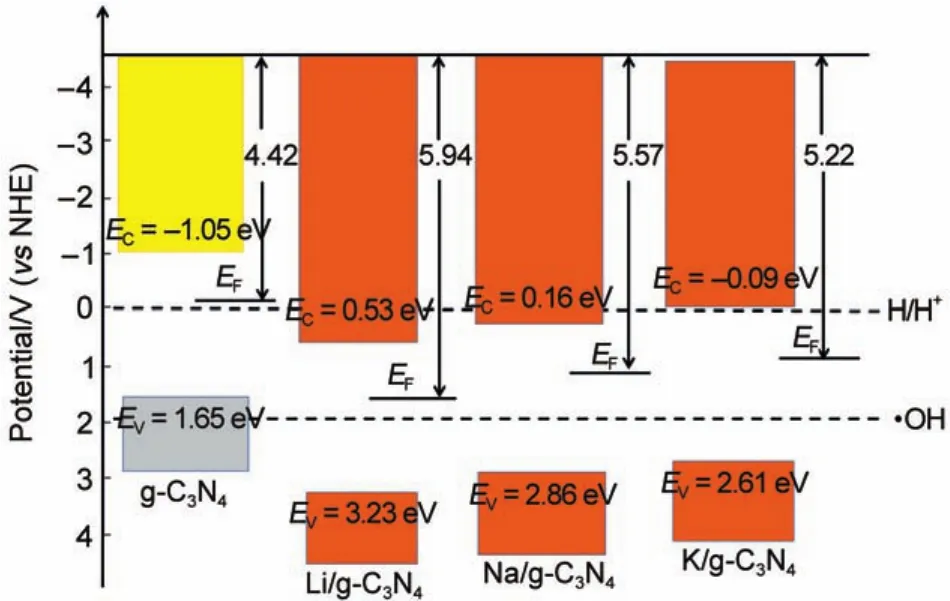
圖3 純g-C3N4和堿金屬摻雜后g-C3N4的導帶和價帶電位Fig.3 Valence band and conduction band potentials for pure g-C3N4and alkali metal doped g-C3N4
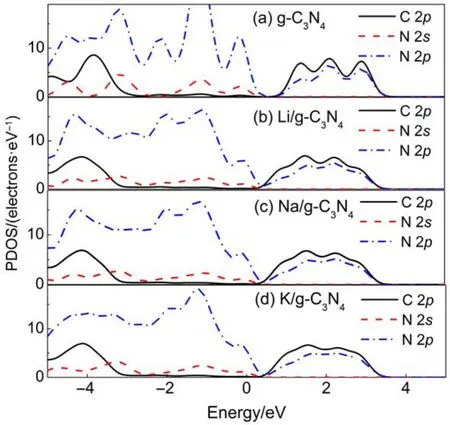
圖4 純g-C3N4和堿金屬摻雜后g-C3N4的分波態密度圖Fig.4 Partial density of states(PDOS)for pure g-C3N4and alkali metal doped g-C3N4
為了進一步分析復雜的活性位置,提供了價帶頂和導帶底附近的HOMO和LOMO信息,如圖5所示。從分布圖來看,摻雜前后的HOMO均主要由低配位N原子在面內的2p軌道構成,而LOMO均主要由C原子在Z方向的2p軌道構成。因此,在可見光激發下空穴僅僅出現在低配位N原子周圍,在面內表現出氧化活性,而電子出現在C原子周圍,在Z方向上表現出還原性。對于純g-C3N4,一方面,HOMO在低配位N原子以及LOMO在C原子Z方向的局域性都較強;另一方面,橋位N原子連接了三個三均三嗪(C6N7)結構單元,它的2p占據態位于費米能級下方0.60 eV位置,未占據態位于導帶底上方1.35 V位置,表明橋位N原子的軌道分布對價帶頂和導帶底沒有任何貢獻,而且局域性很強。也就是說載流子不能通過橋位N原子從一個大三角形的三均三嗪(C6N7)結構單元到其他的結構單元,載流子的遷移受到橋位N原子的限制。
這里以K摻雜為例說明載流子的遷移過程。由于堿金屬的雜質能級位置很深,摻雜后堿金屬離子并不直接參加光生載流子的產生、分離和轉移過程。比較圖5(a)和(b)可以發現,K摻雜后,一方面使橋位N原子附近LUMO的非局域性有所增強,可見光激發下光生電子可以通過橋位的N原子在相鄰的三均三嗪(C6N7)結構單元間自由遷移,這將克服橋位N原子對載流子遷移限制作用,從而提高其遷移率。同時還發現K摻雜使LUMO集中到摻雜層,而HOMO主要集中到非摻雜層。因此,當可見光激發下的光生電子從HOMO躍遷至LUMO,電子和空穴分別在摻雜層和非摻雜層內遷移,大大降低載流子的復合幾率,延長了載流子的壽命。也就是說,增大的HOMO和LOMO分布有利于提高載流子的遷移率,而HOMO和LOMO的非共面特性有利于電子和空穴的分離16,36。

圖5 LUMO和HOMO分布圖Fig.5 Distribution of LUMO and HOMO
4 結論
采用第一性原理方法研究了三種堿金屬離子修飾對g-C3N4能帶結構和載流子遷移過程的影響。對建立的六種吸附構型進行吸附能計算,均發現三種堿金屬原子吸附于g-C3N4片層內的大空洞中央位置(F位置)時,顯示出最大的吸附能且為負數,說明了該吸附構型較易于實現。功函數計算的結果顯示堿金屬與g-C3N4形成的異質結界面的電荷平衡會使g-C3N4的能帶位置整體下移,尤其值得注意的是K離子的引入使g-C3N4的價帶和導帶的電位分別調整為2.51和-0.19 eV,具有比未摻雜的g-C3N4更合適的氧化還原電位。由于堿金屬的雜質能級位置很深,摻雜的堿金屬離子并不直接參加光生載流子的產生、分離和轉移過程,但是影響周圍鄰近其他原子的電荷分布,即表現出HOMO和LOMO非共面特性以及增大的HOMO和LOMO分布,這有利于電子和空穴的分離和遷移。
References
(1) Chen,X.B.;Shen,S.H.;Guo,L.J.;Mao,S.S.Chem.Rev. 2010,110,6503.doi:10.1021/cr1001645
(2) Kudo,A.;Mesiki,Y.Chem.Soc.Rev.2009,38,253. doi:10.1039/B800489G
(3)Zou,Z.G.;Ye,J.H.;Sayama,K.;Arakawa,H.Nature 2001, 414,625.doi:10.1038/414625a
(4) Kudo,A.;Ueda,K.;Kato,H.;Mikami,I.Catal.Lett.1998,53, 229.doi:10.1023/A:1019034728816
(5) Kohtani,S.;Tomohiro,M.;Tokumura,K.;Nakagaki,R.Appl. Catal.B-Environ.2005,58,265.doi:10.1016/j. apcatb.2004.12.007
(6) Fu,H.B.;Zhang,S.C.;Xu,T.G.;Zhu,Y.F.;Chen,J.M.Environ.Sci.Technol.2008,42,2085.doi:10.1021/es702495w
(8) Pan,C.S.;Zhu,Y.F.Environ.Sci.Technol.2010,44,5570. doi:10.1021/es101223n
(9) Ma,X.G.;Lu,B.;Li,D.;Shi,S.;Pan,C.S.;Zhu,Y.F.J.Phys. Chem.C 2011,115,4680.doi:10.1021/jp111167u
(10)Wang,X.C.;Maeda,K.;Thomas,A.;Takanabe,K.;Xin,G.; Carlsson,G.M.;Domen,K.;Antonietti,M.Nat.Mater.2009,8, 76.doi:10.1038/nmat2317
(11) Yan,S.C.;Li,Z.S.;Zou,Z.G.Langmuir 2009,25,10397. doi:10.1021/la900923z
(12) Bojdys,M.J.;Müller,J.;Antonietti,M.Chem.Eur.J.2008,14, 8177.doi:10.1002/chem.200800190
(13) Kroke,E.;Schwarz,M.Coord.Chem.Rev.2004,248,493. doi:10.1016/j.ccr.2004.02.001
(14) Zhang,J.S.;Chen,X.F.;Takanabe,K.Angew.Chem.Int.Ed. 2010,49,441.doi:10.1002/anie.200903886
(15) Liu,G.;Niu,P.;Sun,C.H.J.Am.Chem.Soc.2010,132, 11642.doi:10.1021/ja103798k
(16) Ma,X.G.;Lv,Y.H.;Xu,J.J.Phys.Chem.C 2012,116,23485. doi:10.1021/jp308334x
(17) Zhang,G.G.;Zhang,M.W.;Ye,X.X.;Qiu,X.Q.;Lin,S.; Wang,X.C.Adv.Mater.2014,26,805.doi:10.1002/ adma.201303611
(18) Lin,S.;Ye,X.X.;Gao X.M.;Huang,J.J.Mol.Catal.A:Chem. 2015,406,137.doi:10.1016/j.molcata.2015.05.018
(19) Chen,Y.;Wang,B.;Lin,S.;Zhang,Y.F.;Wang,X.C.J.Phys. Chem.C 2014,118,29981.doi:10.1021/jp510187c
(20) Jin,R.R.;You,J.G.;Zhang,Q.;Liu,D.;Hu,S.Z.;Gui,J.Z. Acta Phys.-Chim.Sin.2014,30,1706.[金瑞瑞,游繼光,張倩,劉丹,胡紹爭,桂建舟.物理化學學報,2014,30,1706.] doi:10.3866/PKU.WHXB201406272
(21) Gao,H.L.;Yan,S.C.;Wang,J.J.Appl.Catal.B-Environ.2014, 158,321.doi:10.1016/j.apcatb.2014.04.036
(22) Zhang,M.;Bai,X.J.;Liu,D.;Wang,J.;Zhu,Y.F.Appl.Catal. B-Environ.2015,164,77.doi:10.1016/j.apcatb.2014.09.020
(23) Segall,M.D.;Lindan,P.L.D.;Probert,M.J.J.Phys.Condens. Matter 2002,14,2717.doi:10.1088/0953-8984/14/11/301
(24) Kohn,W.;Sham,L.J.Phys.Rev.1965,140,A1133. doi:10.1103/PhysRev.140.A1133
(25) Ortmann,F.;Bechstedt,F.;Schmidt,W.G.Phys.Rev.B 2006, 73,205101.doi:10.1103/PhysRevB.73.205101
(27) Deifallah,M.;McMillan,P.F.;Cora,F.J.Phys.Chem.C 2008, 112,5447.doi:10.1021/jp711483t
(28) Pan,H.;Zhang,Y.W.;Shenoy,V.B.;Gao,H.J.ACS Catal. 2011,1,99.doi:10.1021/cs100045u
(29) Gracia,J.;Kroll,P.J.Mater.Chem.2009,19,3013. doi:10.1039/B821568E
(30) Hu,S.Z.;Li,F.Y.;Fan,Z.P.;Wang,F.;Zhao,Y.F.;lv,Z.B. Dalton Trans.2015,44,1084.doi:10.1039/C4DT02658F
(31) Xiong,T.;Cen,W.L.;Zhang,Y.X.;Dong,F.ACS Catal.2016, http://pubs.acs.org on March 7,2016.
(32)Aspera,S.M.;David,M.;Kasai,H.Jap.J.Appl.Phys.2010, 49,115703.doi:10.1143/JJAP.49.115703
(33) Xu,Y.;Schoonen,M.A.A.Am.Mineral.2000,85,543.
(35) Chun,W.J.;Ishikawa,A.;Fujisawa,H.;Takata,T.;Kondo,J. N.;Hara,M.;Kawai,M.;Matsumoto,Y.;Domen,K.J.Phys. Chem.B 2003,107,1798.doi:10.1021/jp027593f
(36) Fukuzumi,S.;Kotani,H.;Ohkubo,K.;Ogo,S.;Tkachenko,N. V.;Lemmetyinen,H.J.Am.Chem.Soc.2004,126,1600. doi:10.1021/ja038656q
Band Structure Modulation and Carrier Transport Process of g-C3N4Doped with Alkali Metals
ZHU Lin1MAXin-Guo1,2,*LIU Na1XU Guo-Wang1,2HUANG Chu-Yun1,2,*
(1School of Science,Hubei University of Technology,Wuhan 430068,P.R.China;2Hubei Collaborative Innovation Center for High-efficiency Utilization of Solar Energy,Hubei University of Technology,Wuhan 430068,P.R.China)
The effects of Li,Na,and K alkali metal ions on the band structures and carrier transfer of graphitic carbon nitride(g-C3N4)are investigated using the plane-wave ultrasoft pseudopotential method.The generalized gradient approximation and local density approximation are used to calculate total energies of six adsorption configurations.The three alkali ions all tend to adsorb on the large central cavity(F position)in g-C3N4layers. The calculated band structures and work function values indicate that the interface charge balance of the n-type Schottky junctions formed between the alkali metal ions and g-C3N4induces the total band edge potential of g-C3N4to shift down by 1.52 V(Li),1.07 V(Na),and 0.86 V(K).The incorporation of K ion adjusts the valence and conduction bands to more appropriate redox potentials than those of pure g-C3N4,and increases the distribution of the HOMO and LOMO of g-C3N4,which helps to improve the mobility of carriers.Meanwhile,the non-coplanar HOMO and LOMO favor the separation of electrons and holes.
Photocatalysis;g-C3N4;Band structure;Carrier transport
March 18,2016;Revised:June 21,2016;Published online:June 22,2016.
s.MAXin-Guo,Email:maxg2013@sohu.com.HUANG Chu-Yun,Email:chuyunh@163.com.
O649
10.3866/PKU.WHXB201606222
The project was supported by the National Natural Science Foundation of China(51102150,51472081),Foundation of Hubei University of
Technology for High-level Talents,China(GCRC13014),Development Founds of Hubei Collaborative Innovation Center,China
(HBSKFZD2014003,HBSKFZD2014011,HBSKFZD2015004),and Students Research Fund of Hubei Collaborative Innovation Center,China (HBSDY201511).
國家自然科學基金(51102150,51472081),湖北工業大學高層次人才啟動基金(GCRC13014),湖北省協同創新中心開放基金項目
(HBSKFZD2014003,HBSKFZD2014011,HBSKFZD2015004)和湖北省協同創新中心大學生科技創新基金(HBSDY201511)資助?Editorial office ofActa Physico-Chimica Sinica
(7)Maeda,K.;Domen,K.Chem.Mater.2010,22,612. 10.1021/cm901917a
(26) Vanderbilt,D.Phys.Rev.B 1990,41,7892.10.1103/ PhysRevB.41.7892
(34) Pearson,R.G.Inorg.Chem.1988,27,734.10.1021/ ic00277a030
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
