Pt/Y催化劑催化FCC柴油加氫制備BTX
2016-11-18 06:47:44周立坤于海斌葛慶峰范景新裴仁彥臧甲忠南軍
化工學報 2016年11期
關鍵詞:催化劑
周立坤,于海斌,葛慶峰,范景新,裴仁彥,臧甲忠,南軍
?
Pt/Y催化劑催化FCC柴油加氫制備BTX
周立坤1,2,于海斌1,葛慶峰2,范景新1,裴仁彥1,臧甲忠1,南軍1
(1中海油天津化工研究設計院有限公司,天津 300131;2天津大學化工學院,天津 300072)
利用Pt/Y催化劑,在固定床反應器中,溫度380℃、壓力3 MPa、氫油體積比1000及質量空速1.0 h-1條件下,分別采用加氫處理的全餾分和輕餾分催柴為原料制備苯、甲苯和二甲苯(BTX),獲得(C6+C7+C8)芳烴的總選擇性分別為9.4%和33.9%。對原料和液體產物進行的氣相色譜和質譜分析表明,BTX主要經過重芳烴的加氫飽和、裂解等反應生成,中間物質為烷基苯、四氫萘、茚滿及茚類等單環芳烴。通過對反應原料以及對反應前后催化劑的N2吸脫附、NH3-TPD、XRD衍射圖譜、TG等物化性質的表征,分析催化劑失活的主要原因。即全餾分催柴原料中高含量的S、N化合物快速吸附造成了催化劑中毒,而輕餾分原料中S、N化合物在催化劑表面的緩慢積累覆蓋活性位,造成催化劑逐漸失活。
催化裂化柴油;重芳烴;分子篩;催化劑;固定床;加氫;BTX
引 言
隨著石油資源的不斷開發消耗,催化裂化柴油(催柴)呈現劣質化、重質化趨勢,其中的芳烴化合物含量也不斷提高。目前,催柴主要作為燃料使用,工業價值相對較低,這也造成了資源的嚴重浪費[1-2]。另外,催柴加氫過程會大大提高生產成本,而用于調和成品油的方案不僅會降低柴油十六烷值,又會加重環境污染。因此,催柴的高值化、環保化利用成為各煉化企業亟待解決的問題。
苯、甲苯和二甲苯(BTX)在石油化工領域中有著重要的作用,是石油化工行業重要的基本原料[3-5]。目前,國內BTX的生產主要以催化重整石腦油,或蒸汽裂化石腦油的副產物為原料獲得[6],二甲苯也可通過芳烴異構化[7]、甲苯歧化和C9芳烴烷基轉移等方法制備[8],生物質經熱分解制備BTX也有探索性研究[9]。國內BTX產能不足,大量依賴進口。因此,以催柴為原料,利用其富含芳烴的特點制備BTX等高附加值化學品的研究被人們逐漸重視。在分子篩擔載的過渡金屬、合金或其氧化物催化劑作用下,重芳烴可通過加氫裂解(開環或脫烷基)、烷基轉移以及異構化等過程生成BTX[10-14]。催柴中含有大量的雙環、多環芳烴類C10+重芳烴。因此,以催柴為原料,在一定條件及催化劑作用下,通過催化加氫裂解等過程可有效提高催柴制備BTX的效率。
加氫裂解催化劑是由具有加氫功能的金屬組分和酸性功能的酸性載體組成[15]。載體的酸性是影響催化裂解的主要因素,能顯著降低裂解反應的溫度。烴類進行裂解、脫烷基的過程是在催化劑的酸性中心上進行的[16-17]。Toppi等[18]根據Pt/SiO2、Pt-Sn/SiO2、Al2O3和Cl-Al2O3催化正丙苯加氫反應得到的產物分布狀況及進行相關動力學研究,對催化劑各種酸中心及金屬中心的作用進行了闡明。他們認為,烴類在強B酸位依據碳正離子機理進行裂解脫烷基反應,在弱B酸中心上主要進行的是異構化過程,強弱L酸上主要是通過自由基過程進行脫烷基反應,在金屬中心上發生的脫氫和氫解過程,遵循吸附脫氫-活性位基元反應機理。在加氫裂解催化劑的作用下,芳烴的輕質化過程也伴隨有烷基轉移反應的發生,受催化劑的酸性位及載體的空間結構影響。此外,氫氣可促進裂解反應的發生,有助于降低裂解溫度。Serra等[19-20]利用不同酸性分子篩分別擔載Re、Pt、Mo、Ga、Ni、La、Bi等金屬制備了幾種催化劑,并用于考察重整油的脫烷基和烷基轉移過程與載體的關系。分子篩的孔徑和結構不僅影響脫烷基過程,對不同烷基的轉移過程也有著直接的影響。如隨著分子篩孔徑的減小,乙基和丙基的脫烷基速率增加,而烷基轉移速率降低。另外,具有較強加氫活性的金屬組分的引入,也會對烷基轉移過程的選擇性產生影響[21]。
影響加氫裂解催化劑失活的原因包括催化劑中毒[17,22-23]、積炭[22]等,其中引起中毒失活的原因又包括吸附和化學反應[22]。由于各種烴類在催化劑上的吸附能力強弱不同,大分子、稠環芳烴的吸附能力更強[24],因此造成的吸附及催化劑孔道堵塞現象更嚴重,容易引起催化劑中毒失活。Kondoh等[17]在利用TiO2-ZrO2催化劑催化重油大分子制備輕型油品的研究中指出,催化劑中晶格氧對C—C鍵斷裂過程提供主要的酸性位。但催化劑上吸附的有機物量會隨反應時間的延長而增加,并逐漸降低其活性。黃星亮等[23]研究了有機硫化物對催化劑活性的影響,指出不同硫化物會在催化劑上發生S—H或S—S鍵斷裂并與催化劑金屬中心形成配合物,致使催化劑活性降低。李文慧[22]對甲苯甲醇烷基化過程ZSM-5催化劑上的積炭物種進行了質譜定性分析,包括多苯、萘、菲、并四苯等多種化合物。說明積炭過程發生了小分子的聚合過程。
Y分子篩具有超籠結構和良好的酸性位,適于催化重芳烴大分子裂解[16-17,25-27]。本文選擇Y型分子篩,通過浸漬法擔載Pt制備的Pt/Y催化劑,利用固定床在氫氣氣氛下通過一步法由催柴制備BTX。結合雙環芳烴的加氫飽和特點,對含重芳烴混合油為原料的輕質化過程可提供借鑒作用。
1 實驗部分
1.1 實驗裝置
實驗裝置如圖1所示,主要由進料進氣組件、固定床反應器、溫度及壓力控制系統、產品分離收集器等部分組成。其中,溫度通過反應器的上、中、下3部分進行監測和控溫。催化劑于常壓下進行還原,還原后在氫氣氣氛下降溫至反應溫度待進料。料液通過泵在一定泵速下進入系統,通過進氣閥和排空閥調節系統壓力,原料油和氫氣順向由固定床反應器頂端進入,與氫氣在催化劑作用下發生反應。包括氣相和液相的混合產物經過氣液分離罐分離后,液體產物流入收集罐待取出分析。
1.2 原料
加氫處理的全餾分催柴WD(以下稱“全餾分催柴”)和再經精餾制得的輕餾分催柴LD(以下稱“輕餾分催柴”)由山東齊旺達集團石油化工有限公司提供,加氫處理的全餾分催柴WD′(以下稱“金陵全餾分催柴”)來源于金陵石化公司。
1.3 反應評價與產物分析
催柴催化轉化制備BTX的評價實驗在固定床反應器中進行,催化劑用量12 g。反應溫度380℃、反應壓力3 MPa、氫油體積比1000、質量空速1.0 h-1。進料前,催化劑在常壓、氫氣氣氛中程序升溫至450℃還原1 h,氫氣流量為100 ml·min-1。原料及液相產物采用Agilent Technologies公司7890B型氣相色譜儀定量分析,色譜柱為HP-5毛細管柱(30 m× 0.32 mm × 0.25 μm),FID檢測。對原料及液相產物的定性分析通過Thermo Scientific公司Trace1310型氣相質譜儀完成,色譜柱為DB-5MS毛細管柱(30 m× 0.25 mm × 0.25 μm)。
1.4 分析測試儀器
氣相色譜分析儀(GC),7890B GC System,美國Agilent Technologies;氣相色譜-質譜聯用儀(GC-MS),TRACE 1310 Gas Chromatograph-ISQ LT Single Quadrupole Mass Spectrometer,美國Thermo SCIENTIFIC;物理吸附儀(Physisorption Analyzer),ASAP 2010,美國Micromeritics;化學吸附儀(Chemisorption Analyzer),AutoChem Ⅱ,美國Micromeritics;原位X射線衍射(XRD-)及PTC-EVO溫度控制器,X-RAY DIFFRACTOMETER,日本Rigaku;X射線熒光光譜分析(XRF),ZSX PrimusⅡ,日本Rigaku;熱重-差熱分析(TG-DTA),METTLER TOLEDO,瑞士;硫氮元素分析儀(GC),MultiTekTM,美國ANTEK。
1.5 數據處理方法
液體產物收率()以及產物中C10+轉化率(10+)分別用式(1)、式(2)計算
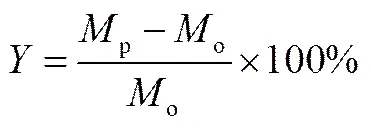
(2)
2 實驗結果與討論
2.1 原料
幾種催柴原料油的密度、硫氮含量、模擬蒸餾的分析結果見表1。由表1可見,全餾分催柴經過精餾切割后,組分中S、N含量明顯降低,分別由原來的432.7×10-6、37.3×10-6 g·cm-3降至20.6×10-6、2.7×10-6 g·cm-3。說明經過加氫處理的催柴中S、N主要集中在高沸點化合物中。模擬蒸餾結果顯示,氣化99.5%的輕餾分催柴所需的溫度為325.6℃,大大低于全餾分催柴的401.6℃。溫度達到121.0℃后,輕餾分催柴氣化量為0.5%,而相同氣化量的全餾分催柴只需37.2℃。這說明,對全餾分催柴進行精餾截取輕組分的過程,窄化了催柴的組分沸點,簡化了其成分組成。S、N含量的降低及高沸點組分的去除使輕餾分催柴的密度減小。
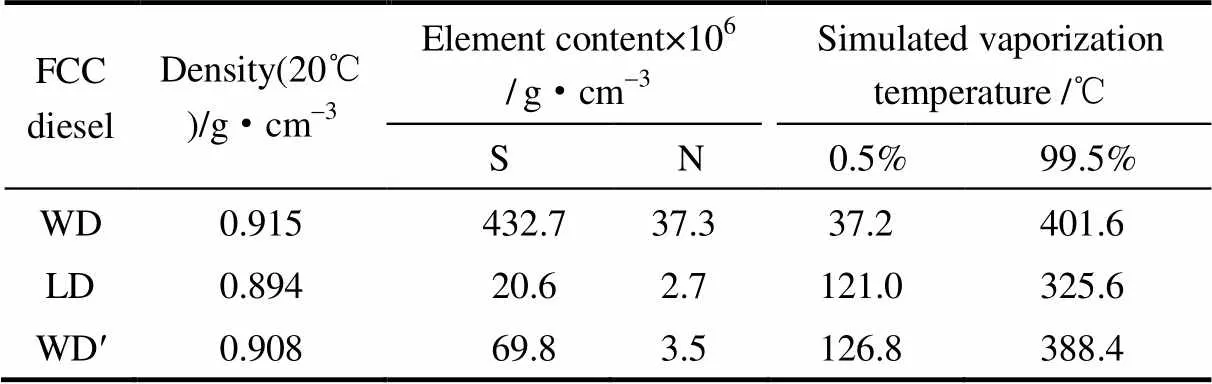
表1 原料油的性質
2.2 催化劑
Y型分子篩與Pt/Y浸漬劑(使用前需經還原處理)由天津正達科技有限責任公司催化劑廠提供,Pt擔載量為0.1%。用于催化全餾分與輕餾分催柴使用后的待生劑,以及經焙燒處理的再生劑分別計作Pt/Y (W-Used)、Pt/Y (L-Used)、Pt/Y (W-Used-CA)與Pt/Y (L-Used-CA)。
載體及催化劑的物理化學性質表征結果如表2所示,Y分子篩和Pt/Y浸漬劑的比表面大小相近,約為500 m2·g-1。使用后的催化劑,比表面積和孔體積都大大降低,用于催化全餾分和輕餾分催柴轉化的催化劑比表面積分別降至5 m2·g-1和30 m2·g-1,總孔容積分別降至0.03 cm3·g-1和0.08 cm3·g-1。通過NH3-TPD測量的酸含量表明,Y型分子篩酸量為0.76 mmol·g-1,負載Pt及經過氫氣還原后略增至0.79 mmol·g-1。使用后的催化劑酸量明顯減少,分別降至0.28 mmol·g-1和0.29 mmol·g-1。催化劑酸含量的減少會直接降低其裂化活性。催化劑使用后經空氣焙燒后,比表面積、孔容及酸含量等物性明顯恢復。對于Pt/Y (W-Used)、Pt/Y (L-Used)催化劑的熱重分析可見,二者失重率分別為28.9%和24.7%,說明催化劑使用后吸附或積炭明顯,用于全餾分催柴轉化的催化劑失重率更大。
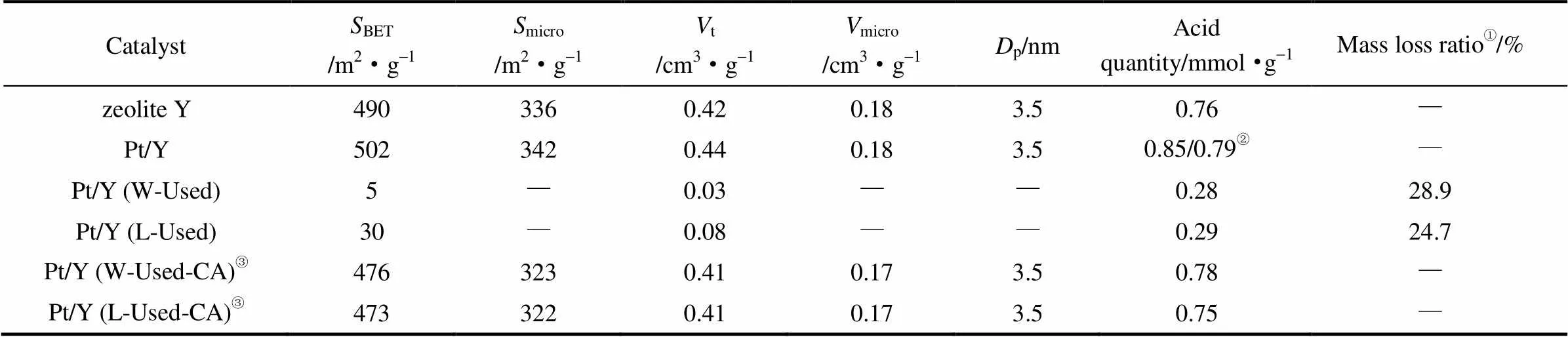
表2 催化劑及載體的物理化學性質
① Used catalysts were calcinated from room temperature to 700℃ at rate of 10℃·min-1and air atmosphere.
② Measured after reducing Pt/Y catalyst at 450℃ for 1 h with 100 ml·min-1H2flow.
③Catalyst was calcinated at 550℃ and air atmosphere for 4 h to constant mass.
Note:BET,micro,t,microandpweremeasured by N2-physisorption. Acid quantity and mass loss were determined by NH3-chemisorption and TG respectively.
載體及催化劑的N2吸脫附等溫曲線如圖2所示。在相對壓力達到0.4以上時,載體及各種催化劑的N2吸脫附等溫曲線有回滯環出現,但沒有明顯的飽和吸附平臺,說明孔結構很不規整。其中,載體zeolite Y,催化劑Pt/Y、Pt/Y (W-Used-CA)、Pt/Y (L-Used-CA)在較高相對壓力吸附曲線升高緩慢,趨于吸附飽和。根據IUPAC分類,這4種催化劑的回滯環屬于H4型,說明載體和催化劑為微孔和中孔混合型。使用后的催化劑經過550℃空氣焙燒處理,孔結構可恢復。而反應后的催化劑Pt/Y (W-Used)、Pt/Y (L-Used)吸附量在較高相對壓力區間內仍隨相對壓力的升高而迅速增大,說明N2的吸附過程未達到飽和狀態。這兩種催化劑的回滯環屬于H3型,反映的孔情況類似于平板狹縫結構等。此外,反應后的催化劑N2的吸附量大大降低,說明有機物吸附在催化劑載體的孔道中發生后,改變了孔結構及孔體積等物理性質。
a—zeolite Y;b—Pt/Y;c—Pt/Y (W-Used);d—Pt/Y (L-Used);e—Pt/Y (W-Used-CA);f—Pt/Y (L-Used-CA)
載體及催化劑的NH3-TPD程序升溫脫附曲線如圖3所示。Zeolite Y、Pt/Y、Pt/Y (W-Used-CA)、Pt/Y (L-Used-CA)的NH3-TPD曲線上存在兩個脫附峰。低溫峰(194℃)代表弱酸位吸附氨的脫附,強酸位吸附氨的脫附出現在358℃高溫峰處。與zeolite Y相比,載體擔載Pt后,弱酸峰和強酸峰都向高溫方向較小遷移。用于催化輕餾分催柴的催化劑經過焙燒處理后弱酸峰和強酸峰出現的位置與新鮮催化劑相當,說明酸強度未有明顯改變,但總酸量降低(表 2)。用于催化全餾分催柴的催化劑經過焙燒處理后弱酸峰和強酸峰都向低溫方向明顯遷移。原因可能是,全餾分催柴中較高的S含量(表 1)在反應過程中改變了催化劑的酸性位,從而弱化了催化劑的酸強度。從圖中可以明顯看到,使用后的催化劑Pt/Y (W-Used)、Pt/Y (L-Used)只在弱酸區有NH3的脫附峰,較新鮮催化劑相比,弱酸峰向低溫方向明顯遷移。可見,反應過程中有機物的吸附沉積,大大降低了催化劑的酸強度及酸量,而強酸位受影響更大。
a—zeolite Y;b—Pt/Y;c—Pt/Y (W-Used);d—Pt/Y (L-Used);e—Pt/Y (W-Used-CA);f—Pt/Y (L-Used-CA)
載體與催化劑的XRD衍射圖及Pt/Y (L-Used)催化劑的原位XRD衍射圖如圖4所示。從圖4(A)可見,分別用于催化全餾分和輕餾分催柴轉化的Pt/Y (W-Used)(c)、Pt/Y (L-Used)(d)催化劑,晶面(1 1 1)和(2 2 0)處衍射峰強度減弱,經過空氣焙燒后恢復載體以及新鮮催化劑的衍射峰強度。在對Pt/Y (L-Used)催化劑進行原位升溫XRD測試過程中,空氣氣氛下從室溫開始,溫度每升高50℃記錄一次樣品XRD衍射圖。從圖4(B)的結果可清楚看見,隨著溫度的升高,晶面(1 1 1)和(2 2 0)處衍射峰強度逐漸增強。空氣及高溫條件下,催化劑孔道內積聚的碳氫化合物逐漸分解,骨架中的硅鋁結構逐漸暴露,穩定性提高,相對結晶度也逐漸恢復。
2.3 氣相色譜分析結果
全餾分、輕餾分催柴原料和相應液相產物的氣相色譜分析結果如圖5所示。
全餾分催柴作為反應底物時[圖5(a)],液體產物的收率達到90%以上。液相產物中C5化合物、C6~C9芳烴等單環烴類、非芳化合物的相對含量隨反應時間的變化趨勢類似,呈現先增加后減小再至平穩的過程。反應24 h后C8和C9芳烴的選擇性最大,分別達到液體產物的6.6%和8.7%。產物中C10+、C12+化合物的相對含量隨時間呈現先減少后增加再至平穩的變化趨勢,與單環化合物的變化呈互補的關系,C10+、C12+化合物含量在24 h最低,分別為75%和46%。可以認為,產物中的單環化合物的生成主要通過C10+化合物的裂解反應實現。隨著催化劑在使用過程中逐漸失活,對C10+化合物的裂解能力逐漸降低,C10+轉化率也隨反應時間的延長而逐漸減小。
輕餾分催柴作為反應底物時[圖5(b)],與全餾分催柴作為填料的實驗結果類似,液相產物中C5化合物、C6~C9芳烴等單環化合物的相對含量隨反應時間的變化趨勢與C10+、C12+化合物呈互補的關系,同樣說明單環化合物的生成主要來源C10+等化合物。與全餾分催柴為原料的反應相比[圖5(a)],反應0~24 h內C10+、C12+化合物的含量減少明顯,分別減少了81.4%和69.2%。但C5化合物、C6~C9芳烴等單環化合物增量較小,增量最多的為C8芳烴,只有15.2%。而此時液體的收率只有66.8%,C10+化合物轉化率達到89.9%,可見反應初始,大部分C10+化合物在裂解過程生成了氣態產物。隨著反應時間的延長,催化劑活性位減少,裂解生成氣體產物減少,液體收率在反應進行96 h后接近90%,并隨反應時間的延長逐漸遞增。同樣,C10+化合物的轉化率隨催化劑使用時間的延長而逐漸降低,反應432 h后降至36.9%。
由此可見,以輕餾分催柴為原料時,催化劑的催化裂解活性降低較小,失活速度較慢。對比催柴原料的S、N含量表征結果(表1),可以認為,高含量的硫、氮化合物的吸附沉積導致催化全餾分催柴催化劑活性位減少,酸量降低(表2、圖3),裂化活性降低。含硫、氮化合物的性質可導致催化劑的活性位銳減,如有機堿性含氮化合物可與催化劑上的酸性位點發生強烈的化學吸附,使酸性中心中毒[28]。含硫化合物通過硫分子在催化劑活性位的吸附和氫解過程,可與催化劑的金屬組分生成穩定的Me—S非活性硫化物[29]。
新鮮催化劑在反應起始階段活性變化明顯,因此,在0~56 h時間內縮短取樣時間,考察產物變化情況。實驗考察了氫氣氣氛下Y分子篩對反應的催化效果,反應以加氫處理的金陵全餾分催柴為原料,產物色譜分析結果見圖6。
Y分子篩作催化劑使用時對應產物的分布以及隨反應時間的變化趨勢[圖6(a)]與Pt/Y催化劑[圖6(b)]相近。負載Pt后,相同時間點催化劑表現出更強的裂解能力,液體收率更低,反應2 h時只有25%,此時C10+轉化率為85.6%,為所有檢測點的最大值。而Pt的引入明顯提高了催化劑的加氫活性,對應的非芳含量增加,反應8 h時非芳含量達到56.2%,對應的Y分子篩作為催化劑時,非芳含量為20.6%。由于重芳烴輕質化過程主要包括C—C鍵裂解和加氫兩類反應,因此Pt/Y催化劑具有的酸性和加氫功能分別提供催化兩種反應的活性位,二者在催化過程中呈現協同作用。此外,短時間記錄的產品變化規律與長時間取樣產物分布結果對比可進一步確定,新鮮催化劑隨反應時間的延長,催化劑吸附有機物引起中毒等原因造成了活性位點的減少,使得催化劑活性降低[17,22-24]。
催柴在Pt/Y催化劑作用下發生裂解等反應,生成多種單環化合物,其中,苯、甲苯、二甲苯為關注的高附加值化合物。雖然C8芳烴也包含乙苯,但是乙苯同樣為附加值較高的化學品,因此,本文主要計算C6、C7、C8等芳烴的總含量。圖7給出了全餾分和輕餾分催柴制備BTX反應中,產物(C6+C7+C8)芳烴的總選擇性。
由圖可見,全餾分催柴為原料的反應,(C6+C7+C8)芳烴總選擇性低于9.4%,隨著反應的進行,總選擇變化較小(反應432 h后,值為7.1%)。輕餾分催柴為底物時,(C6+C7+C8)芳烴總選擇性在反應48 h后達到最大值33.9%。隨著反應時間的延長,選擇性大大降低,至反應進行432 h后,其值降到11.7%。(C6+C7+C8)芳烴總選擇性隨反應時間的延長減少明顯,原料大分子吸附[24]、含硫含氮化合物引起的催化劑中毒[23],以及反應中積炭過程使催化劑活性位不斷減少[30-31],致使催化劑的加氫裂解能力逐漸降低,相應的催化劑表征見表2、圖2~圖4。輕餾分催柴在精餾處理后S、N含量降低(表1),緩解了催化劑因吸附含硫、氮物質而中毒失活。但不易發生加氫反應的含氮化合物積累到一定程度時,同樣會吸附在催化劑表面而影響催化劑的裂化和加氫性能的匹配。而含硫化合物在催化劑表面活性中心的吸附達到一定程度時,同樣可引起催化劑積炭的產生[28]。
對比用于全餾分和輕餾分催柴催化劑的反應結果,前者失活更快,待生劑上有機物吸附量也更多(表2)。另外,由于全餾分催柴中的大分子烴類含量更多,因此,造成的吸附及催化劑孔道堵塞現象更嚴重,用于催化此原料的催化劑失活更快。
2.4 質譜分析結果
催柴原料在反應溫度380℃、壓力3 MPa、氫油體積比1000以及空速為1.0 h-1條件下,及催化劑Pt/Y作用下,反應24 h后和反應432 h后產物的質譜分析結果如表3所示。
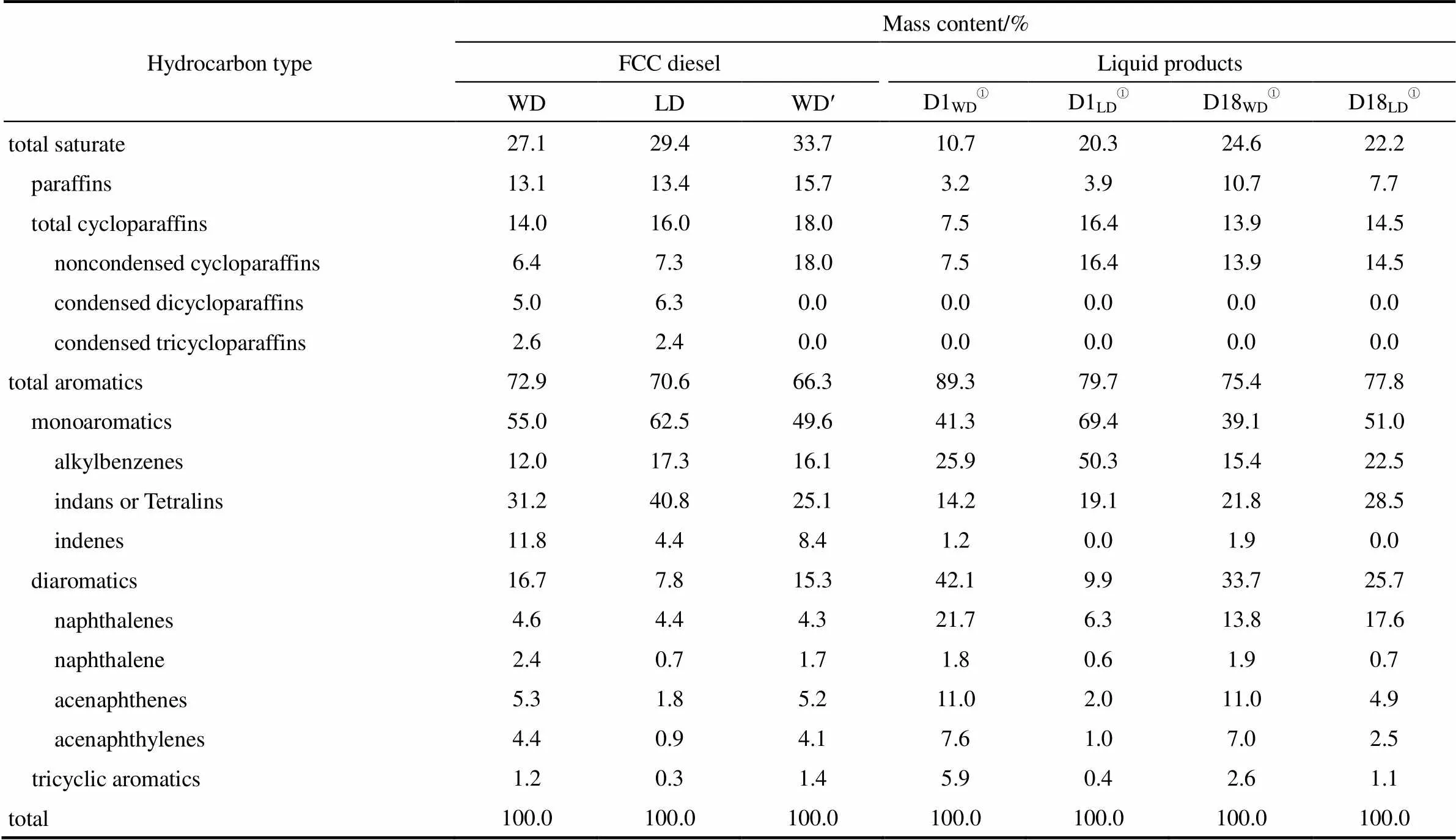
表3 原料油及液相產物的質譜分析
①D1WD,D1LD,D18WDandD18LD: productsfrom catalyst serving 1 d or 18 d for WD and LD feed, respectively.
Note: Reaction conditions:=380℃;=3 MPa;(H2)/(Feed)=1000; WHSV=1.0 h-1.
催柴原料的質譜分析表明,由全餾分催柴經切割獲得的輕餾分,總飽和烴、總芳烴的含量都沒有較大變化。但輕餾分催柴中單環芳烴的含量百分比升高(由全餾分催柴的55.0%升至62.5%),但雙環芳烴和三環芳烴相對含量減少。說明對催柴進行的加氫精餾處理不僅降低了其中的硫氮含量(表1),同時也改變了其中的芳烴組成。
分別以全餾分和輕餾分催柴為底物時,反應24 h產物組成變化明顯。液體產物中總飽和烴含量減少,尤其是鏈烷烴和雙、三環烷烴相對含量降低明顯。對全餾分和切割后輕餾分催柴的反應,鏈烷烴相對含量分別由原料的13.1%、13.4%降至3.2%、3.9%。雙、三環烷烴的相對含量都減少到0,即使是反應432 h后,二者值仍是0。對于單環烷烴相對含量的變化,反應開始后的值較原料增加。以全餾分原料為反應物時,產物中單環烷烴的相對含量隨反應時間的增加而增大(反應24 h后單環烷烴的含量為7.5%,反應432 h后值為13.9%)。在催柴制備BTX的過程中,引起產物中雙、三環烷烴的減少,單環烷烴相對增加的可能原因,是反應條件下雙、三環烷烴先裂解成單環烷烴。于珊等[32]利用環己烷和十氫萘在研究環數對加氫裂解的影響中得到,相同反應條件下,十氫萘(雙環)較環己烷(單環)的轉化率高。認為可能的原因是,裂解過程中十氫萘形成了叔正碳離子,而環己烷形成的是仲正碳離子,根據雙分子裂化反應機理,前者較后者更容易生成。由表中數據可看到,產物中的鏈烷烴相對含量同樣由于發生裂解反應而減少,隨著反應時間的延長,其含量所占比重升高。另外,可以發現,鏈烷烴的含量減少量較環烷烴相比更大。以輕餾分催柴為底物的反應為例,反應432 h后產物中的鏈烷烴和環烷烴的含量分別減少了5.7%和1.5%。這說明,反應條件下,鏈烷烴較環烷烴更易發生裂解反應。對比反應原料及反應24 h和反應432 h產物的總飽和烴的含量,可見其變化趨勢與液體產物的收率變化趨于一致,說明催化劑的加氫裂解能力降低,活性位減少。
全餾分和輕餾分催柴分別為底物時,反應24 h后產物中總飽和烴相對含量分別由原料的27.1%、29.4%減少到10.7 %和20.3%;總芳烴相對含量分別由原料的72.9%、70.6%提高至89.3%和79.7%。飽和烴相對含量的減少,以及芳烴相對含量增加的現象,可以被認為是,反應條件下飽和烴較芳烴更易發生裂解反應引起的。在芳烴產物中,除萘以外的雙環、總雙環芳烴以及三環芳烴的相對含量增加,除烷基苯外的茚滿、四氫萘等單環及總單環芳烴相對含量減少。另外,雖然雙環芳烴萘的含量相對減少,但萘在催柴中的含量較少(在全餾分和輕餾分催柴中分別占2.4%和0.7%)。因此,可以認為包括BTX等烷基苯芳烴化合物的增加主要是由茚滿、四氫萘以及茚類等單環芳烴的裂解引起的。這與色譜分析得到的結論一致(圖5),即C6~C9芳烴等單環化合物主要來源C10+化合物的裂解反應。同樣,隨著反應時間的延長,催化劑逐漸失活,烷基苯芳烴的增加量減少。如對于輕餾分催柴底物來說,較原料相比,反應24 h后產物中烷基苯相對含量增加33.0%,而反應432 h后烷基苯相對增加量降到5.2%。
對催柴原料及產物的質譜分析結果總結可知,反應條件下,BTX等烷基苯化合物主要由催柴中烷基苯、茚滿、四氫萘以及茚類等帶有飽和環的芳烴催化轉化而得。
2.5 催柴制備BTX的路徑
催柴的成分復雜,其中的單環、雙環及多環等芳烴理論上都可以通過裂解、異構化、烷基轉移等反應過程生成BTX等烷基苯。通過上面的質譜分析可知,在實驗條件下,目標產物主要來自催柴中茚滿、四氫萘等脂環族芳香烴成分。有文獻報道[33],四氫萘在加氫裂化生成小分子的過程中,通常會發生3種反應(圖8):(1)脫氫生成萘;(2)芳環加氫先生成十氫萘,經過進一步的異構、裂解反應生成小于C10的烷烴;(3)飽和六元環經異構化生成1-甲基茚滿,之后裂化為丁基苯,進一步裂化則生成取代基更小的烷基苯。而主導四氫萘加氫反應進行的方向主要受催化劑的種類、反應溫度等條件影響。本實驗條件下,四氫萘生成BTX烷基苯等液相產物的過程,主要通過反應(3)路徑實現。
重芳烴來源廣泛,各組分組成及含量不盡相同,因此,在輕質化過程中需要圍繞反應路徑來設計工藝路線及催化劑。重芳烴中萘及烷基萘等多芳烴化合物在加氫條件下,先經過選擇性加氫(非深度加氫)生成單環飽和芳烴四氫萘及苯并環烷烴等,此過程所需要的反應溫度及催化劑等與加氫裂化最佳反應條件不同。通常,前者在較低反應溫度及Ni基催化劑作用下即可實現。Ren等[34]利用Ni/MMT-CTAB催化劑催化萘選擇加氫制備四氫萘,在300℃反應溫度下,萘的轉化率為88.2%,四氫萘選擇性為99.4%。萘催化加氫過程中,升高反應溫度有利于十氫萘生成[35],因此,過高的反應溫度不利于重芳烴輕質化中間物種四氫萘的生成,進而會降低目標產物BTX的收率。四氫萘異構生成甲基茚滿的活化能也高于萘的非飽和過程[33]。綜上可得,以含有萘及烷基萘等C10+重芳烴為原料制備BTX過程經歷:(1)低溫加氫生成四氫萘及苯并環烷烴等飽和單環芳烴[34,36];(2)高溫加氫裂化、異構生成BTX,催化劑活性組分選用與C9+制備BTX過程中的Pt系貴金屬催化劑[37-39]。
輕質化過程中催化劑的載體選擇、改性是一項關鍵問題,包括酸性和孔結構。酸性過強易造成過度裂化,降低液收。催化劑的孔道尺寸不僅影響反應分子的傳質過程,同時較大分子在孔口處容易堵塞而引起催化劑失活[40-41]。因此,適宜的孔道能為反應物提供更多的活性位。此外,對于雙環及多環芳烴含量不多的重芳烴原料,可先通過精餾處理對單環芳烴進行富集,能大大降低原料中大分子吸附造成的中毒失活,對提高輕質化催化劑的穩定性具有促進作用。
3 結 論
在Y型分子篩擔載的Pt催化劑、反應溫度380℃、壓力3 MPa、氫油體積比1000、空速1.0 h-1條件下,由全餾分或輕餾分催柴制備BTX的過程,得到如下結論。
(1)對加氫處理后的全餾分催柴進行精餾處理獲得的輕餾分催柴,S、N含量大大降低,茚類及雙、三環芳烴等重組分相對含量也明顯減少。
(2)催化加氫全餾分催柴為原料的反應,(C6+C7+C8)芳烴總選擇性低于9.4%,但隨著反應時間的延長,總選擇性減小緩慢。輕餾分催柴為底物時,(C6+C7+C8)芳烴總選擇性的最大值達到33.9%,總選擇性隨反應時間的延長而降低明顯。
(3)催化全餾分催柴的催化劑活性低的主要原因是,反應開始時原料中高含量的S、N化合物快速吸附使催化劑中毒。催化輕餾分催柴的催化劑逐漸失活的主要原因是,原料中含S、N化合物在催化劑表面的緩慢積累覆蓋了活性位,此外,反應過程產生的積炭也會造成催化劑失活。
(4)對全餾分和輕餾分催柴轉化的液體產物氣相色譜分析可知,C5化合物、C6~C9芳烴等單環化合物主要來源于C10+化合物的裂解反應。
(5)BTX等烷基苯化合物主要由催柴中烷基苯、茚滿、四氫萘以及茚類等單環芳烴轉化而得。
(6)以C10+為原料的輕質化工藝,可結合低溫非深度加氫與高溫催化裂解制備BTX。
符 號 說 明

C10+——C10+轉化率,% Dp——孔平均尺寸,nm Mo——進料質量,g Mp——液體產物質量,g SBET——比表面積,m2·g-1 Smicro——微孔面積,m2·g-1 Vmicro——微孔體積,cm3·g-1 Vt——總孔體積,cm3·g-1 Xo(10+)——原料中C10+含量,% Xp(10+)——產物中C10+含量,% Y——液體產物收率,% 下角標 o——原料 p——液體產物 10+——分子中碳數≥10
References
[1] 申群兵, 朱學棟, 董嬌嬌, 等. 載鉬分子篩催化劑上C9+重芳烴的輕質化[J]. 石油學報(石油加工), 2009, 25(3): 351-357. SHEN Q B, ZHU X D, DONG J J,. Conversion of C9+heavy aromatics to light aromatics over molybdenum-based zeolite catalysts[J]. Acta Petrol. Sin(Pet. Process Section), 2009, 25(3): 351-357.
[2] 董嬌嬌, 朱瑾, 申群兵, 等. MoO3/HZSM-5催化劑上重芳烴加氫脫烷基反應[J]. 石油化工, 2008, 37(3): 232-237. DONG J J, ZHU J, SHEN Q B,. Hydrodealkylation of heavy aromatics on MoO3/HZSM-5 catalyst[J]. Petrochem. Techno., 2008, 37(3): 232-237.
[3] SHEN Q B, ZHU X D, DONG J J,. Hydrodealkylation of C9+heavy aromatics to BTX over zeolite-supported nickel oxide and molybdenum oxide catalysts[J]. Catal. Lett., 2009, 129(1/2): 170-180.
[4] 戴厚良. 芳烴生產技術展望[J]. 石油煉制與化工, 2013, 44(1): 1-10. DAI H L. Outlook of aromatics production technology[J]. Chin. Petrol. Processing Petrochem. Technol., 2013, 44(1): 1-10.
[5] TSAI T C, LIU S B, WANG I. Disproportionation and transalkylation of alkylbenzenes over zeolite catalysts[J]. Appl. Catal. A-Gen., 1999, 181(2): 355-398.
[6] SUN C, SEUNG H O, YONG S K,. APUSMtechnology for the production of BTX and LPG from pyrolysis gasoline using metal promoted zeolite catalyst[J]. Catal. Surv. Asia, 2009, 10(2): 110-116.
[7] 李麗娟, 劉君. 芳烴異構化過程的多模型建模[J]. 化工學報, 2011, 62(8): 2350-2354. LI L J, LIU J. Multi-modeling of aromatics isomerization process[J]. CIESC Journal, 2011, 62(8): 2350-2354.
[8] 徐歐官, 蘇宏業, 金曉明, 等. 用于甲苯歧化與C9芳烴烷基轉移過程先進控制的反應動力學模型[J]. 化工學報, 2007, 58(3): 630-637. XU O G, SU H Y, JIN X M,. A kinetic model for advanced process control of toluene disproportionation and transalkylation with C9-aromatics[J]. Journal of Chemical Industry and Engineering (China), 2007, 58(3): 630-637.
[9] 王昶, 賈青竹, 中川紳好, 等. 木材經催化熱分解向BTX和合成燃料的轉化[J]. 化工學報, 2004, 55(8): 1341-1347. WANG C, JIA Q Z, ZHONGCHUAN S H,. Conversion of plant-biomass to BTX and synthetic fuels by catalytic pyrolysis[J]. Journal of Chemical Industry and Engineering(China), 2004, 55(8): 1341-1347.
[10] 楊平, 辛靖, 李明豐, 等. 四氫萘加氫轉化研究進展[J]. 石油煉制與化工, 2011, 42(8): 1-6. YANG P, XIN J, LI M F,. Research advances in the hydroconversion of tetralin[J]. Chin. Petrol. Processing Petrochem. Technol., 2011, 42(8): 1-6.
[11] 孔德金, 祁曉嵐, 朱志榮, 等. 重芳烴輕質化技術進展[J]. 化工進展, 2006, 25(9): 983-987. KONG D J, QI X L, ZHU Z R,. Technological advances in conversion of heavy aromatics to light aromatics[J]. Chem. Ind. Eng. Prog., 2006, 25(9): 983-987.
[12] 唐之勤, 王德舉. 重整重芳烴催化加氫裂解反應工藝條件的探究[J]. 化學反應工程與工藝, 2011, 27(1): 1-5. TANG Z Q, WANG D J. Process conditions for catalytic hydropyrolysis of heavy reformates[J]. Chem. React. Eng. Technol., 2011, 27(1): 1-5.
[13] 季靜, 柴忠義. 重質芳烴輕質化技術研究進展[J]. 化學工業, 2013, 31(4): 25-27. JI J, CHAI Z Y. The research process of heavy aromatic to lightweight technology[J]. Chemical Industry, 2013, 31(4): 25-27.
[14] 唐占忠, 陳一齋. 由C9+重芳烴制取輕芳烴[J]. 現代化工, 1994, (1): 20-30. TANG Z Z, CHEN Y Z. Preparing light aromatic hydrocarbons from C9+heavy aromatic hydrocarbons[J]. Mod. Chem. Ind., 1994, (1): 20-30.
[15] 張健, 魯海龍, 孫建偉, 等. 分子篩和γ-Al2O3混合載體上負載Ni和W的加氫裂解催化劑的EXAFS研究[J]. 高等學校化學學報, 2000, 21(10): 1459-1463. ZHANG J, LU H L, SUN J W,. EXAFS study of Ni/W/zeolite/Al2O3hydrocracking catalyst sulfurized with different modified zeolites[J]. Chem. J. Chin. Univ., 2000, 21(10): 1459-1463.
[16] ZHAO J, WANG G G, QIN L H,. Synthesis and catalytic cracking performance of mesoporous zeolite Y[J]. Catal. Commun., 2016, 73: 98-102.
[17] KONDOH H, TANAKA K, NAKASAKA Y,. Catalytic cracking of heavy oil over TiO2-ZrO2catalysts under superheated steam conditions[J]. Fuel, 2016, 167: 288-294.
[18] TOPPI S, THOMAS C, SAYAG C,. Kinetics and mechanisms of-propylbenzene hydrodealkylation reactions over Pt(Sn)/SiO2and (Cl-)Al2O3catalysts in reforming conditions[J]. J. Catal., 2002, 210(2): 431-444.
[19] SERRA J M, GUILLON E, CORMA A. Optimizing the conversion of heavy reformate streams into xylenes with zeolite catalysts by using knowledge base high-throughput experimentation techniques[J]. J. Catal., 2005, 232(2): 342-354.
[20] SERRA J M, GUILLON E, CORMA A. A rational disign of alkyl-aromatics dealkylation-transalkylation catalysts using C8and C9alkyl-aromatics as reactants[J]. J. Catal., 2004, 227(2): 459-469.
[21] TSAI T C, CHEN W H, LIU S B,. Metal zeolites for transalkylation of toluene and heavy aromatics[J]. Catal. Today, 2002, 73(1/2): 39-47.
[22] 李文慧. ZSM-5分子篩催化劑上積碳物種的研究[D]. 大連: 大連理工大學, 2014. LI W H. The study of coke species on ZSM-5 zeolite catalysts[D]. Dalian: Dalian University of Technology, 2014.
[23] 黃星亮, 沈師孔. 有機硫化物使Pd/樹脂催化劑中毒的規律與機理[J]. 催化學報, 2003, 24(3): 233-237. HUANG X L, SHEN S K. Effect of organic sulfur compounds on isoprene-hydrogenation reactivity over Pd/resin and its poisoning mechanism[J]. Chin. J. Catal., 2003, 24(3): 233-237.
[24] 劉銀東, 李澤坤, 王剛, 等. 競爭吸附對催化裂化反應過程的影響[J]. 化工學報, 2008, 59(11): 2794-2799. LIU Y D, LI Z K, WANG G,. Effect of competitive adsorption on catalytic cracking reaction[J]. Journal of Chemical Industry and Engineering(China), 2008, 59(11): 2794-2799.
[25] DU X H, LI X L, ZHANG H T,. Kinetics study and analysis of zeolite Y destruction[J]. Chin. J. Catal., 2016, 37(2): 316-323.
[26] WARD J J, LINDA Y. Hydrocracking catalyst and process: US5350501[P]. 1994-09-27.
[27] ZE?EVI? J, GOMMES C J, FRIEDRICH H,. Mesoporosity of zeolite Y: quantitative three-dimensional study by image analysis of electron tomograms[J]. Angew. Chem. Int. Edit., 2012, 51(17): 4213-4217.
[28] 張喜文, 馬波, 凌鳳香. 原料油中氮、硫體積分數及反應壓力對加氫裂化催化劑積炭的影響[J]. 燃料化學學報, 2005, 33(1): 101-106. ZHANG X W, MA B, LING F X. Effects of nitrogen or sulphide content and reaction pressure on coke deposited on hydrocracking catalyst[J]. J. Fuel Chem. Technol., 2005, 33(1): 101-106.
[29] 侯朝鵬, 李永丹, 趙地順. 芳烴加氫金屬催化劑抗硫性研究的進展[J]. 化工進展, 2003, 22(4): 366-371. HOU C P, LI Y D, ZHAO D S. Research on sulfur tolerance of metal catalysts in aromatics hydrogenation[J]. Chem. Ind. Eng. Prog., 2003, 22(4): 366-371.
[30] 王玉忠, 張立新, 李秀杰, 等. PtSnZn/ZSM-5 -ZSM-11-Al2O3催化劑上FCC汽油制苯、甲苯和二甲苯[J]. 石油化工, 2010, 39(6): 625-630. WANG Y Z, ZHANG L X, LI X J,. Aromatization of FCC gasoline to BTX aromatics over PtSnZn/ZSM-5 -ZSM-11-Al2O3catalysts[J]. Petrochem. Techno., 2010, 39(6): 625-630.
[31] 陳賢峰. C8芳烴在Oparis催化劑上的臨氫異構化反應過程建模和催化劑失活研究[D]. 上海: 華東理工大學, 2014. CHEN X F. Process modeling of C8-aromatics hydroisomerization reaction over oparis bifunctional catalyst and study of the inactivation of catalyst[D]. Shanghai: East China University of Science and Technology, 2014.
[32] 于珊, 張久順, 魏曉麗. 環烷烴催化裂解生成乙烯和丙烯反應探析[J]. 石油學報(石油加工), 2013, 29(3): 475-481. YU S, ZHANG J S, WEI X L. Exploration and analysis on ethylene and propylene formation in naphthene catalytic cracking[J]. Acta Petrol. Sin(Pet. Process Section), 2013, 29(3): 475-481.
[33] 宋欣. 雙環化合物加氫裂化反應規律研究[D]. 青島: 中國石油大學(華東), 2007. SONG X. A study on hydrocracking reaction of bicyclic compound over zeolite catalysts[D]. Qingdao: China University of Petroleum , 2007.
[34] REN S B, WEN H Z, CAO X Z,. Promotion of Ni/clay catalytic activity for hydrogenation of naphthalene by organic modification of clay[J]. Chin. J. Catal., 2014, 35(4): 546-552.
[35] 夏良燕, 夏芝香, 方夢祥, 等. 煤焦油中芳烴(萘)的加氫飽和試驗[J]. 浙江大學學報(工學版), 2015, 49(3): 578-584. XIA L Y, XIA Z X, FANG M X,. Hydrogenation saturation of aromatic compounds (naphthalene) in coal tar[J]. Journal of Zhejiang University(Engineering Science), 2015, 49(3): 578-584.
[36] PAWELEC B, CASTANO P, ARANDES J M,. Factors influencing the thioresistance of nickel catalysts in aromatics hydrogenation[J]. Appl. Catal. A-Gen., 2007, 317(1): 20-33.
[37] HOWLEY P A, SHIH S S. Catalytic hydrodealkylation of aromatics: US5001296[P]. 1991-03-19.
[38] WU A H, DRAKE C A. Hydrodealkylation catalyst composition and process therewith: US5698757[P]. 1997-12-16.
[39] WU A H, DRAKE C A, MELTON R J. Catalyst composition and processes therefore and therewith: US5714660[P]. 1998-02-03.
[40] 陳振濤, 徐春明. 重質油在孔道內擴散傳質的研究進展[J]. 化工學報, 2016, 67(1): 165-175. CHEN Z T, XU C M. Progress of research on diffusional transport of heavy oil in pores[J]. CIESC Journal, 2016, 67(1): 165-175.
[41] HAUSER A, MARAFI A, ALMUTAIRI A,. Comparative study of hydrodemetallization (HDM) catalyst aging by Boscan feed and Kuwait atmospheric residue[J]. Energ. Fuel , 2008, 22(5): 2925-2932.
Synthesisof BTX by Pt/Y catalyzed hydrogenation of FCC diesel
ZHOU Likun1,2, YU Haibin1, GE Qingfeng2, FAN Jingxin1, PEI Renyan1, ZANG Jiazhong1, NAN Jun1
(1Cener Tech Tianjin Chemical Research and Design Institute Limited Liability Company, Tianjin 300131, China;2College of Chemical Engineering, Tianjin University, Tianjin 300072, China)
BTX was prepared by Pt/Y catalyzed hydrogenation of the whole or light fraction of hydrogenated FCC diesel in fixed bed reactor at reaction condition of temperature at 380℃, pressure at 3 MPa, volume ratio of(H2) over(feed) at 1000, and WHSV at 1.0 h-1. The overall selectivity of (C6+C7+C8) arenes was 9.4% and 33.9% for the whole and light fraction ofthe hydrotreated FCC diesel, respectively. GC-MS study on feeds and liquid products showed that BTX were mainly produced through hydrogenation saturation and cracking of heavy arenes along with intermediate products of mononuclear aromatics such as alkylbenzenes, indane, tetralin, and indenes. By analysis of feeds, fresh and used catalystsN2-physisorption, NH3-TPD, XRD, and TG, it was found that adsorption of high sulfur- and nitrogen- compounds in the whole fraction of FCC diesel quickly lead to catalyst toxication whereas slow accumulation and active site coverage on catalyst surface by sulfur- and nitrogen- compounds in the light fraction of FCC diesel gradually lead to catalyst deactivation.
FCC diesel; heavy aromatics; molecular sieves; catalyst; fixed bed; hydrogenation; BTX
2016-06-08.
10.11949/j.issn.0438-1157.20160795
TQ 028.8
A
0438—1157(2016)11—4623—11
劣質柴油生產芳烴工業技術開發(中海油能源發展股份有限公司科技項目)。
2016-06-08收到初稿,2016-08-31收到修改稿。
聯系人及第一作者:周立坤(1982—),男,博士,工程師。
ZHOU Likun, lincoln_chou@yeah.net
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
