多次溶劑萃取-氣相色譜/串聯(lián)質(zhì)譜法測(cè)定熱熔膠中的多環(huán)芳烴
2016-11-09 08:32:47司曉喜朱瑞芝張鳳梅劉春波徐艷群李中昌何沛劉志華
分析化學(xué) 2016年3期
司曉喜 朱瑞芝 張鳳梅 劉春波 徐艷群 李中昌 何沛 劉志華
(云南省煙草化學(xué)重點(diǎn)實(shí)驗(yàn)室,云南中煙工業(yè)有限責(zé)任公司技術(shù)中心,昆明650231)
多次溶劑萃取-氣相色譜/串聯(lián)質(zhì)譜法測(cè)定熱熔膠中的多環(huán)芳烴
司曉喜朱瑞芝張鳳梅劉春波徐艷群李中昌何沛劉志華*
(云南省煙草化學(xué)重點(diǎn)實(shí)驗(yàn)室,云南中煙工業(yè)有限責(zé)任公司技術(shù)中心,昆明650231)
建立了熱熔膠中16種多環(huán)芳烴(PAHs)的多次溶劑萃取-氣相色譜/串聯(lián)質(zhì)譜測(cè)定方法。詳細(xì)研究了樣品的萃取條件、凈化條件和氣相色譜/串聯(lián)質(zhì)譜測(cè)定條件,并與氣相色譜-質(zhì)譜法進(jìn)行了對(duì)比。樣品以10 mL正己烷為萃取溶劑,于60℃超聲萃取20min,萃取液依次經(jīng)冷凍后離心、二甲基亞砜萃取2次、正己烷反萃取2次進(jìn)行凈化,得到的凈化液以氣相色譜/串聯(lián)質(zhì)譜法多反應(yīng)監(jiān)測(cè)(MRM)模式進(jìn)行檢測(cè)。本方法的線性相關(guān)系數(shù)(R2)均大于0.9969,檢出限為1.0~10μg/kg,精密度小于6.3%,16種PAHs的加標(biāo)回收率為80.4%~117.6%。考察了串聯(lián)質(zhì)譜檢測(cè)的基質(zhì)效應(yīng),發(fā)現(xiàn)基質(zhì)效應(yīng)不明顯。本方法檢出限優(yōu)于氣相色譜-質(zhì)譜法(23~94μg/kg),并能增加定性和定量分析的準(zhǔn)確性。本方法靈敏、準(zhǔn)確可靠,滿足熱熔膠中PAHs測(cè)試要求。
多環(huán)芳烴;溶劑萃取;氣相色譜-串聯(lián)質(zhì)譜;熱熔膠
1 引言
熱熔膠是以熱塑性樹(shù)脂或彈性體為基料,添加增黏劑、增塑劑、抗氧化劑、阻燃劑及填料,經(jīng)熔融混合而成的固體狀粘合劑[1]。熱熔膠廣泛應(yīng)用于各領(lǐng)域,但其主體原料乙烯-醋酸乙烯聚合物、石蠟等均為石化產(chǎn)品,存在著多環(huán)芳烴(PAHs)污染的可能性[2,3],增粘劑“氫化石油樹(shù)脂”中也可能存在芳香單體殘留[4]。多環(huán)芳烴具有致癌、致畸和致突變的作用,對(duì)人類健康和生態(tài)環(huán)境具有巨大的潛在危害[5]。歐盟2002/72/EC指令中規(guī)定了“氫化石油樹(shù)脂”中殘留芳香單體≤50 mg/kg,GB 7189-2010采用“紫外吸光度”限量的方法控制食品級(jí)石蠟中PAHs含量。盡管對(duì)熱熔膠的一些組成原料有PAHs的限量要求,但目前我國(guó)還沒(méi)有熱熔膠中PAHs的限量要求和檢測(cè)方法,因此建立熱熔膠中PAHs的檢測(cè)方法對(duì)熱熔膠的安全性研究十分必要。
目前,測(cè)定環(huán)境樣品、塑料制品中PAHs的報(bào)道較多[6],常用方法主要是紫外吸光光度法和氣相色譜-質(zhì)譜聯(lián)用法[7]。GBT 7363-1987采用紫外吸光光度法測(cè)定石蠟中PAHs的紫外吸光度,操作繁瑣,且不能得到PAHs的含量。氣相色譜-質(zhì)譜法快速、靈敏度高,但是抗基質(zhì)干擾能力一般[8]。氣相色譜-串聯(lián)質(zhì)譜法(GC-MS/MS)靈敏度高,選擇性好,十分適合復(fù)基質(zhì)中痕量污染物的檢測(cè)[9~11]。此外,熱熔膠性狀特別、基質(zhì)復(fù)雜[12~14],需要經(jīng)過(guò)特殊的前處理才能進(jìn)行分析。常用的PAHs提取方法有索氏提取法、微波萃取法、超聲萃取法等;凈化方法主要有固相萃取法和液液萃取法[15]。其中微波萃取法、超聲萃取法快速、萃取效率高;固相萃取法集樣品富集及凈化于一體,但可能造成目標(biāo)物的損失,液液萃取法簡(jiǎn)便,對(duì)性質(zhì)差異較大的干擾物去除效果好[15]。
本研究針對(duì)熱熔膠樣品的特殊性,擬開(kāi)發(fā)集成超聲萃取和液液萃取凈化的多次溶劑萃取前處理方法,并采用氣相色譜/串聯(lián)質(zhì)譜測(cè)定熱熔膠中16種PAHs。
2 實(shí)驗(yàn)部分
2.1儀器、試劑與材料
Scion TQ氣相色譜-串聯(lián)質(zhì)譜儀,配456 GC氣相色譜儀和自動(dòng)進(jìn)樣器(美國(guó)布魯克·道爾頓公司);超聲波清洗器(昆山市超聲儀器有限公司),配有溫控功能,功率≥200 W;BT224S電子天平(感量0.0001 g,德國(guó)賽多利斯科學(xué)儀器有限公司);3K15高速臺(tái)式離心機(jī)(德國(guó)Sigma公司,);旋轉(zhuǎn)蒸發(fā)儀(瑞士Buchi公司);Milli-Q超純水器(美國(guó)Millipore公司)。
正己烷、環(huán)己烷(色譜純,美國(guó)J.T.Baker公司);二甲基亞砜(色譜純,德國(guó)Merk公司);NaCl(分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司);16種多環(huán)芳烴混合標(biāo)樣,包括萘(NAP)、苊烯(ANY)、苊(ANA)、芴(FLU)、菲(PHE)、蒽(ANT)、熒蒽(FLT)、芘(PYR)、苯并(a)蒽(BaA)、(CHR)、苯并(b)熒蒽(BbF)、苯并(k)熒蒽(BkF)、苯并(a)芘(BaP)、茚苯(1,2,3-cd)芘(IPY)、二苯并(a,h)蒽(DBA)、苯并(g,h,i)芘(BPE),濃度均為1 mg/mL(美國(guó)O2si公司,);氘代萘-D8(內(nèi)標(biāo)物1)、氘代蒽-D10(內(nèi)標(biāo)物2)、氘代苝-D12(內(nèi)標(biāo)物3)(標(biāo)準(zhǔn)品,純度≥99%,德國(guó)Dr.Ehrenstorfer公司)。市售熱熔膠樣品,以及自制添加了16中多環(huán)芳烴的陽(yáng)性樣品。
用正己烷配制濃度為20μg/mL的氘代萘-D8、氘代蒽-D10、氘代苝-D12混合內(nèi)標(biāo)溶液。
用正己烷配制系列濃度為0.01,0.05,0.2,0.5,1.0和2.0 mg/L的16種多環(huán)芳烴混合標(biāo)準(zhǔn)溶液,并加入內(nèi)標(biāo),使得內(nèi)標(biāo)濃度均為1.0 mg/L。
2.2樣品前處理
熱熔膠樣品前處理過(guò)程如圖1所示。
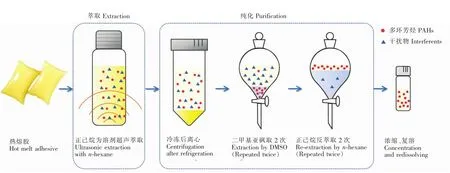
圖1 熱熔膠中多環(huán)芳烴前處理過(guò)程示意圖Fig.1 Diagram of polycyclic aromatic hydrocarbons(PAHs)pretreatment process from hotmelt adhesive DMSO:Dimethyl sulfoxide.
2.2.1樣品萃取將熱熔膠樣品切碎成粒徑小于1 mm的顆粒。準(zhǔn)確稱取切碎的樣品0.25 g置于22 mL頂空瓶,加入10 mL正己烷,同時(shí)加入50μL內(nèi)標(biāo)溶液,密閉,放入超聲波水浴中,于恒溫60℃下萃取20 min,至熱熔膠樣品分散或溶解。萃取液轉(zhuǎn)移至離心管,冷凍10 min,經(jīng)高速離心機(jī)離心5 min。
2.2.2樣品凈化轉(zhuǎn)移上清液至已加入8 mL二甲基亞砜的分液漏斗中,劇烈搖動(dòng)約1 min,離心或靜置分層后,將二甲基亞砜萃取液轉(zhuǎn)移至另一個(gè)分液漏斗中,殘液再用8mL二甲基亞砜重復(fù)提取一次,合并萃取液;向萃取液中加入5 mL正己烷和80 mL 4%NaCl溶液,劇烈搖動(dòng)2 min,靜置分層。將下層水相放入另一個(gè)分液漏斗中,再用5 mL正己烷重復(fù)提取一次,合并萃取液。將萃取液連續(xù)用5 mL 4% NaCl溶液洗滌2次,棄去水層,收集正己烷層,在溫度不大于35℃時(shí),采用旋轉(zhuǎn)蒸發(fā)儀將萃取物濃縮至近干,加入1 mL正己烷復(fù)溶,供測(cè)試。因二甲基亞砜熔點(diǎn)為18℃,以上實(shí)驗(yàn)需在室溫大于20℃下操作。
2.3氣相色譜/串聯(lián)質(zhì)譜分析條件
2.3.1氣相色譜條件DB-5MS毛細(xì)管色譜柱(30 m×0.25 mm×0.25μm,美國(guó)Agilent公司);進(jìn)樣口溫度:280℃;傳輸線溫度:280℃;溶劑延遲:7.5 min;載氣:氦氣(純度≥99.999%),恒流流速:1.5 mL/min;進(jìn)樣量:1μL,分流進(jìn)樣(分流比 10∶1);程序升溫:初始溫度 60℃,保持 1 min,然后以6℃/min升至120℃,再以5℃/min升至300℃,并保持13 min。
2.3.2串聯(lián)質(zhì)譜條件離子源溫度:200℃;四極桿溫度:40℃;電離模式:電子轟擊離子源(EI),電子能量:70 eV;燈絲發(fā)射電流80μA;碰撞氣:高純氬氣(純度≥99.999%),壓力:23.8 Pa;檢測(cè)方法:多反應(yīng)監(jiān)測(cè)模式(MRM),設(shè)定l6種PAHs的檢測(cè)窗口,分別測(cè)定其離子強(qiáng)度。其它質(zhì)譜參數(shù)見(jiàn)表1。

表1 16種多環(huán)芳烴的串聯(lián)質(zhì)譜和單四極桿質(zhì)譜參數(shù)Table 1 Parameters of MS/MSand MS for determination of 16 polycyclic aromatic hydrocarbons(PAHs)
3 結(jié)果與分析
3.1樣品萃取條件優(yōu)化
根據(jù)相似相溶原理以及熱熔膠的性質(zhì),考察了正己烷、二甲基亞砜、二氯甲烷、乙酸乙酯等溶劑對(duì)加標(biāo)陽(yáng)性樣品的萃取效果。其中,二氯甲烷會(huì)使熱熔膠溶脹,萃取液呈粘稠狀,不利于后續(xù)處理與測(cè)定;乙酸乙酯、二甲基亞砜等對(duì)熱熔膠溶解或分散效果差,萃取率低;正己烷對(duì)熱熔膠溶解性好,萃取率高,因此選擇正己烷為萃取劑。
進(jìn)一步比較了萃取方式(加熱回流法、超聲萃取法及微波萃取法)、萃取時(shí)間(10,20,40和60 min)和溶劑用量(5,10,20和40 mL)對(duì)PAHs的萃取效果,以PAHs的平均萃取率作為評(píng)價(jià)指標(biāo),結(jié)果見(jiàn)圖2。由圖2A可見(jiàn),超聲萃取法和微波萃取法的萃取率最高,但微波萃取法較為劇烈,易造成萘等低沸點(diǎn)PAHs的損失,此外萃取液冷卻后,析出的熱熔膠附著在提取罐上,難于清洗去除。超聲萃取法的萃取率高,且簡(jiǎn)單、方便,適合批處理,因此選擇超聲萃取方式。萃取時(shí)間過(guò)長(zhǎng),小分子量的PAHs有一定的損失,選取萃取時(shí)間為20min。萃取劑用量過(guò)少,不能使熱熔膠分散或溶解;萃取劑用量過(guò)大,增加后續(xù)處理工作量,浪費(fèi)溶劑,且易造成PAHs的損失,綜合考慮,選取10 mL萃取劑。60℃下,熱熔膠樣品能較好地溶解或分散,考慮到高溫易造成低沸點(diǎn)的PAHs損失,因此萃取溫度選取60℃。

圖2 萃取方式、萃取時(shí)間(A)和溶劑用量(B)對(duì)熱熔膠中多環(huán)芳烴萃取率的影響Fig.2 Influence of recovery of different extractionmethods,extraction time(A)and volume of solvent(B) on 16 PAHs from hotmelt adhesive
3.2樣品凈化條件優(yōu)化
萃取液中含有大量熱熔膠基體和干擾物,首先采用冷凍后離心的方法,促使大量基體快速析出,通過(guò)離心實(shí)現(xiàn)基體與萃取液分離。結(jié)果表明,上清液中仍殘留有大量來(lái)自石蠟的C17~C36烷烴混合物,其性質(zhì)與PAHs接近,且其分子量恰好覆蓋了分子量在250~280的PAHs,給PAHs的定性與定量分析造成了干擾,因此需要進(jìn)一步選擇并考察凈化方法。
對(duì)比了固相萃取[16]和液液萃取的凈化效果。固相萃取柱凈化后,提取液明顯變得澄清,表明大部分熱熔膠基體被去除,但小分子量的多環(huán)芳烴損失嚴(yán)重,不能滿足分析要求。根據(jù)正構(gòu)烷烴與PAHs在二甲亞砜(DMSO)溶劑中的溶解度差異,考察了液液萃取的凈化效果。首先采用對(duì)PAHs溶解性更強(qiáng)的DMSO萃取上清液中的PAHs,以空白基質(zhì)加標(biāo)樣品進(jìn)行實(shí)驗(yàn),考察了DMSO用量(5,8,10和15 mL)和萃取次數(shù)(1~3次)對(duì)萃取率的影響。由圖3A可見(jiàn),1次萃取難于萃取完全,因此采用少量溶劑多次萃取方式,以8 mL DMSO為萃取劑,重復(fù)萃取2次,多環(huán)芳烴的平均萃取率超過(guò)99.2%,可視為基本萃取完全。采用足量NaCl溶液除去DMSO,并用正己烷反萃取多環(huán)芳烴,考察了正己烷用量(5,10和15 mL)和萃取次數(shù)(1~3次)對(duì)萃取率的影響。由圖3B可見(jiàn),以5 mL正己烷為萃取劑,重復(fù)萃取2次,多環(huán)芳烴的平均萃取率超過(guò)99.7%,基本萃取完全。正己烷萃取液經(jīng)洗滌、濃縮、復(fù)溶,最終得到澄清的萃取液,經(jīng)測(cè)定發(fā)現(xiàn),基質(zhì)干擾顯著降低。
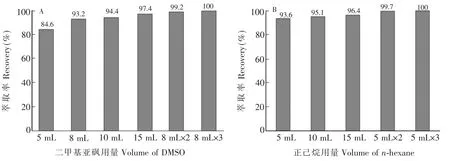
圖3 二甲基亞砜用量(A)和正己烷用量(B)對(duì)多環(huán)芳烴回收率的影響Fig.3 Influence of DMSO volume(A)and n-hexane volume(B)on recovery of 16 PAHs
3.3儀器測(cè)定條件優(yōu)化
熱熔膠樣品提取液經(jīng)處理后仍有少量干擾物,宜采用較緩的升溫程序,使干擾物與目標(biāo)物分離,增加定性與定量分析的準(zhǔn)確性。此外,進(jìn)樣口溫度較高,會(huì)降低小環(huán)PAHs的響應(yīng)強(qiáng)度,增加大環(huán)PAHs的響應(yīng)強(qiáng)度,綜合考慮,進(jìn)樣口溫度選擇280℃。隨著離子源溫度升高,大部分PAHs的響應(yīng)值均有所提高,最終選取離子源溫度為200℃。
以標(biāo)準(zhǔn)溶液進(jìn)行母離子掃描,確定母離子,再進(jìn)行子離子掃描,選取響應(yīng)高、干擾小的兩個(gè)子離子,以MRM對(duì)碰撞能量等質(zhì)譜參數(shù)進(jìn)行優(yōu)化,確定的質(zhì)譜參數(shù)列于表1中。在優(yōu)化的條件下,標(biāo)準(zhǔn)溶液的MRM譜圖見(jiàn)圖4。

圖4 多反應(yīng)監(jiān)測(cè)模式下16種多環(huán)芳烴標(biāo)準(zhǔn)工作溶液的色譜圖Fig.4 Chromatogram of 16 PAHs standard substances undermultiplemonitormode(MRM)化合物序號(hào)同表1。The numbers of compounds are the same as in Table 1.
3.4方法評(píng)價(jià)
取系列混合標(biāo)準(zhǔn)工作液,按優(yōu)化好的儀器條件進(jìn)樣分析。以各PAHs的定量離子峰面積與內(nèi)標(biāo)物定量離子峰面積的比值為縱坐標(biāo),各PAHs的濃度為橫坐標(biāo),繪制標(biāo)準(zhǔn)曲線,內(nèi)標(biāo)法定量。將空白樣品測(cè)定20次,以空白響應(yīng)的3倍標(biāo)準(zhǔn)偏差作為檢出限。將自制陽(yáng)性樣品按照上述實(shí)驗(yàn)方法處理后測(cè)定,以7次平行樣測(cè)定的峰面積計(jì)算相對(duì)標(biāo)準(zhǔn)偏差。取空白樣品,添加含量為0.8,4.0和16.0 mg/kg的各PAHs標(biāo)準(zhǔn)物質(zhì),進(jìn)行加標(biāo)回收實(shí)驗(yàn)(表2)。結(jié)果表明,在所考察濃度范圍內(nèi),各PAHs線性回歸良好,相關(guān)系數(shù)(r2)大于 0.9969。方法的檢出限為 1.0~10μg/kg,檢測(cè)實(shí)際樣品相對(duì)標(biāo)準(zhǔn)偏差小于6.3% (n=7),加標(biāo)回收率為80.4%~117.6%。
3.5基質(zhì)效應(yīng)評(píng)價(jià)
GC-MS/MS測(cè)定復(fù)雜樣品時(shí),基質(zhì)成分可能對(duì)目標(biāo)物響應(yīng)值造成影響,且一般具有增強(qiáng)效應(yīng),影響分析的準(zhǔn)確度和靈敏度[17]。通過(guò)比較純?nèi)軇┡渲频臉?biāo)準(zhǔn)溶液和空白基質(zhì)配制的標(biāo)準(zhǔn)溶液中待測(cè)物的響應(yīng)信號(hào)強(qiáng)度來(lái)評(píng)價(jià)基質(zhì)效應(yīng)。選取1.0μg/mL濃度水平的混合標(biāo)準(zhǔn)溶液進(jìn)行考察,純?nèi)軇┡渲频臉?biāo)準(zhǔn)溶液待測(cè)物響應(yīng)值記為A1,空白基質(zhì)配制的標(biāo)準(zhǔn)溶液待測(cè)物響應(yīng)值記為A2,則基質(zhì)效應(yīng)(ME)= A2/A1。由表2可見(jiàn),16種分析物均表現(xiàn)出較弱的增強(qiáng)效應(yīng),在101.3%~113.4%之間,增強(qiáng)效應(yīng)不明顯,可以忽略。這表明樣品前處理過(guò)程中去除了大部分基質(zhì),減弱了基質(zhì)效應(yīng)[18]。
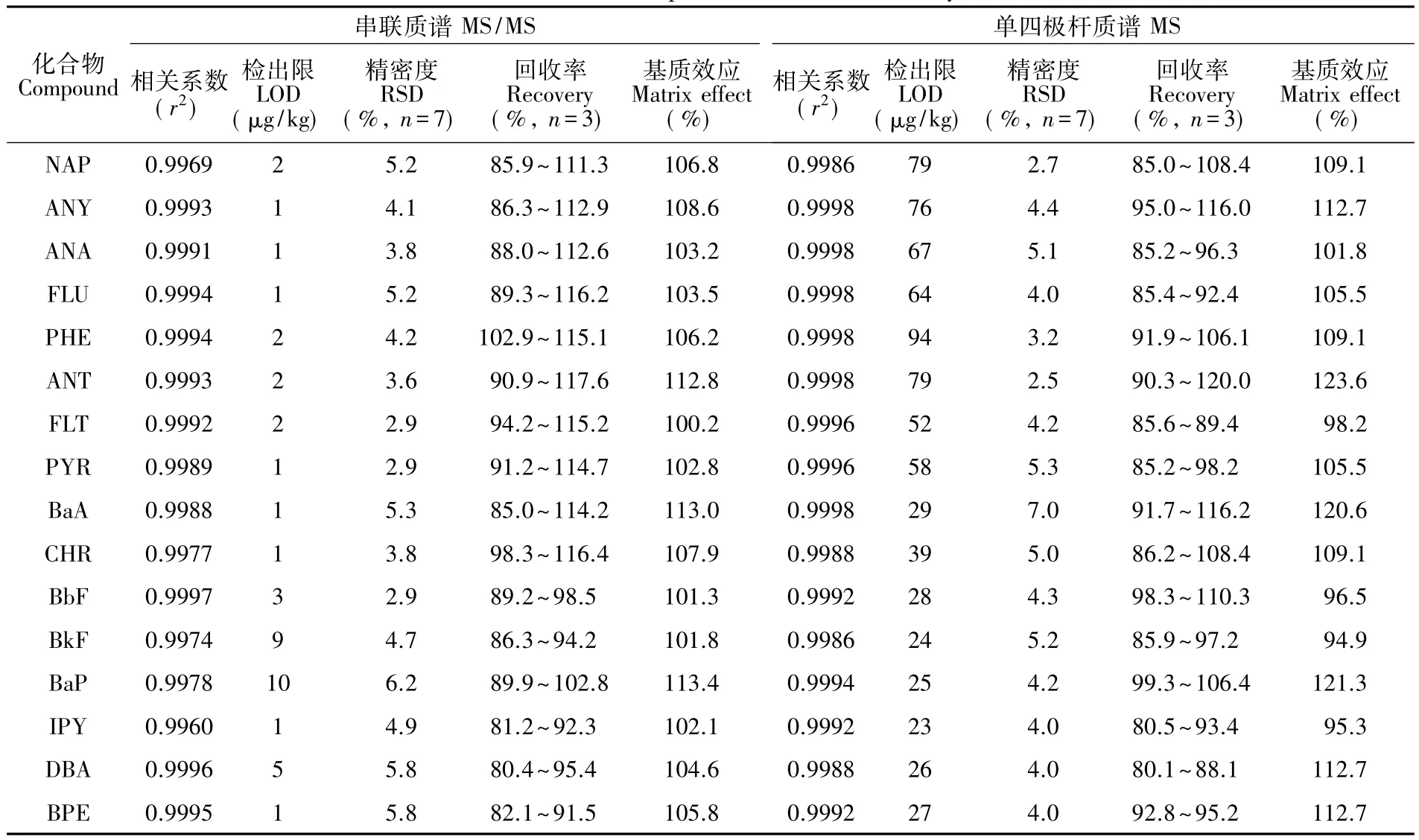
表2 線性相關(guān)系數(shù)、方法的檢出限、精密度和回收率Table 2 Linear correlation coefficient,limit of detection,precision and the recovery test
3.6與氣相色譜-質(zhì)譜法的比較
GC-MS法的線性相關(guān)系數(shù)、檢出限、精密度和回收率見(jiàn)表2。GC-MS/MS法精密度和回收率與GCMS法相當(dāng),但檢出限遠(yuǎn)低于GC-MS法,表明GC-MS/MS法靈敏度明顯優(yōu)于GC-MS法。此外,GC-MS/ MS法選擇性和抗基質(zhì)干擾能力也明顯優(yōu)于GC-MS法。圖5為兩種方法測(cè)定實(shí)際樣品中蒽的離子流圖,GC-MS/MS法信噪比高,兩個(gè)離子對(duì)m/z 178/176、m/z178/152的響應(yīng)比值為100∶86.2,與推測(cè)值100∶86.8十分接近,而GC-MS法中3個(gè)離子m/z178、176、177測(cè)得的豐度比為100∶20∶10,與推測(cè)值100∶14∶8差異較大,可能受干擾物的碎片離子共溢出影響或者基質(zhì)效應(yīng)的影響,容易造成定性與定量分析不準(zhǔn)確。由表2可見(jiàn),GC-MS法的基質(zhì)效應(yīng)在94.9%~123.6%之間,也表明GC-MS法基質(zhì)效應(yīng)稍大于GC-MS/MS法。綜上可知,與GC-MS法相比,GC-MS/MS的定性與定量分析結(jié)果更加準(zhǔn)確。
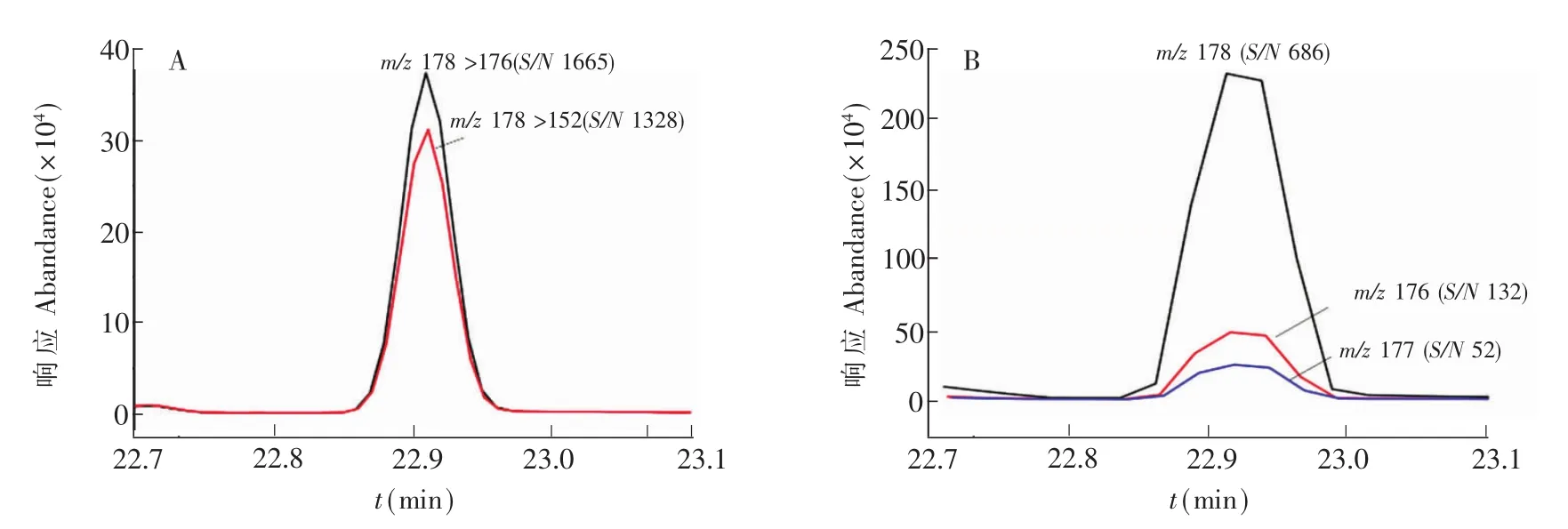
圖5 實(shí)際樣品中蒽的GC-MS/MS測(cè)定MRM離子流圖(A)和GC-MS測(cè)定SIM離子流圖(B)Fig.5 Ion chromatograms of anthracene in sample by GC-MS/MSwith MRM(A)and by GC-MSwith SIM(B)
3.7實(shí)際樣品測(cè)定
采用本方法測(cè)定40個(gè)市售熱熔膠樣品,所測(cè)樣品中有少部分檢出萘、菲和蒽,萘含量為0.08~1.21mg/kg,檢出率為 21.1%;菲含量為 0.08~4.75 mg/kg,檢出率為 23.7%;蒽含量為0.17~4.77mg/kg,檢出率為28.9%,檢出的多環(huán)芳烴總量在0.08~9.63mg/kg之間,存在潛在的風(fēng)險(xiǎn)。
References
1Malysheva G V,Bodrykh N V.Polym.Sci.Ser.D.,2011,4(4):301-303
2Park Y J,Kim H J.Int.J.Adhes.,2003,23(5):383-392
3GUO Jing,XIANG Heng-Xue,WANG Qian-Qian,GUAN Fu-Chen.China Adhesives,2010,19(7):54-58郭靜,相恒學(xué),王倩倩,管福成.中國(guó)膠粘劑,2010,19(7):54-58
4DU Xin-Sheng,YANG Cheng-Jie,ZHANG Lin,XU Hui-Jian,PAN Guang-Qin.ShanghaiCoatings,2013,51(6):37-40杜新勝,楊成潔,張霖,徐惠儉,潘廣勤.上海涂料,2013,51(6):37-40
5HafnerW D,Carlson D L,Hites R A.Environ.Sci.Technol.,2005,39(19):7374-7379
6Poster D L,Schantz M M,Sander L C,Wise SA.Anal.Bioanal.Chem.,2006,386(4):859-881
7Wise SA,Sander L C,Schantz M M.Polycycl.Aromat.Comp.,2015,35(2-4):187-247
8Wang X Y,Wang Y,Qin Y Q,Ding L,Chen Y,Xie FW.Talanta,2015,140:102-108
9Ballesteros E,Sánchez A G,Martos N R.J.Chromatogr.A.,2006,1111(1):89-96
10Strher G L,Poppi N R,Raposo JL,Souza JBG.Microchem.J.,2007,86(1):112-118
11LIU Fei,DUAN Feng-Kui,LIHai-Rong,MA Yong-Liang,HE Ke-Bin,ZHANG Qian.Chinese J.Anal.Chem.,2015,43 (4):540-546劉斐,段鳳魁,李海蓉,馬永亮,賀克斌,張倩.分析化學(xué),2015,43(4):540-546
12CHEN Hai-Yan.China Adhesives,2010,19(5):61陳海燕.中國(guó)膠粘劑,2010,19(5):61
13Khairullin I I,Aleksashin V M,Petrova A P.Polym.Sci.Ser.C.,2007,49(1):84-88
14Park Y J,Joo H S,Kim H J,Lee Y K.Int.JAdhes.Adhes.,2006,26(8):571-576
15ZHANG Ru-Ping,HE Li-Fang.Food science and technology,2007,32(1):20-23章汝平,何立芳.食品科技,2007,32(1):20-23
16MarcéR M,Borrull F.J.Chromatogr.A,2000,885(1):273-290
17SONG Ying,ZHANG Yao-Hai,HUANG Xia,PAN Jia-Rong,JIAO Bi-Ning.Chinese J.Anal.Chem.,2011,39(8):1270-1273宋瑩,張耀海,黃霞,潘家榮,焦必寧.分析化學(xué),2011,39(8):1270-1273
18Frenich A G,Vidal JLM,Moreno JL F,Romero-González R.J.Chromatogr.A,2009,1216(23):4798-4808
Thiswork was supported by the standards project of China National Tobacco Company(No.2013QB015)and the Technological development projects of China Tobacco Yunnan Industrial Co.,Ltd.(No.2015JC06)
Determination of Polycyclic Aromatic Hydrocarbons in Hot Melt Adhesive by Multiple Solvent Extraction Coupled with Gas Chromatography-Tandem Mass Spectrometry
SIXiao-Xi,ZHU Rui-Zhi,ZHANG Feng-Mei,LIU Chun-Bo,XU Yan-Qun,LIZhong-Chang,He Pei,LIU Zhi-Hua*
(Key Laboratory of Tobacco Chemistry,R&D Center of China tobacco Yunnan Industrial Co.,Ltd.,Kunming 650231,China)
Amethod for multiple solvent extraction coupled with gas chromatography-tandemmass spectrometry(GC-MS/MS)was established for the simultaneous determination of 16 polycyclic aromatic hydrocarbons(PAHs)in hotmelt adhesive.The extraction conditions,purification conditions,chromatogphic conditions and tandem MS conditions of analysiswere critically examined.The resultswere also compared with those obtained by gas chromatography-mass spectrometry(GC-MS).The sample was extracted by ultrasonic extraction with n-hexane as solvent at 60℃.Then,the extract was purified by centrifugation after freezing treatment,twice extraction with dimethyl sulfoxide,twice re-extraction with n-hexane in turn.The purified extract was determined by gas chromatography-tandem mass spectrometry undermultiple reaction monitoring (MRM)mode.The linear correlation coefficients were above 0.9969,the limits of detection(LOD)ranged from 1.0μg/kg to 10μg/kg,the relative standard deviations(RSDs)were below 6.3%,and the recoveries ranged from 80.4%to 117.6%at three spiked levels.Thematrix effects of tandem MSwere studied,but there was no significance.Compared with GC-MS methods,the established method outperformed the GC-MS methods in LOD,which ranged from 23μg/kg to 94μg/kg for GC-MS,and could greatly enhance the accuracy of the qualitative and quantitative analysis of PAHs.The results indicated that the method was sensitive,accurate and reproducible,and suitable for the simultaneous determination of 16 PAHs in hotmelt adhesive.
Polycyclic aromatic hydrocarbons;Solvent extraction;Gas chromatography-tandemmass spectrometry;Hotmelt adhesive
17 September 2015;accepted 6 November 2015)
10.11895/j.issn.0253-3820.150739
2015-09-17收稿;2015-11-06接受
本文系中國(guó)煙草總公司標(biāo)準(zhǔn)項(xiàng)目(No.2013QB015)和云南中煙工業(yè)有限責(zé)任公司科技開(kāi)發(fā)項(xiàng)目(No.2015JC06)資助
*E-mail:474629695@qq.com
