基于超高效合相色譜對黃芪中5種主要黃酮類化合物的快速檢測
2016-10-21 11:35:22王波周圍劉小花柳小亞陳亞麗封士蘭
分析化學 2016年5期
王波 周圍 劉小花 柳小亞 陳亞麗 封士蘭
摘要:建立了利用超高效合相色譜法(Ultra performance convergence chromatography,UPC2)快速分離和測定黃芪中5種主要黃酮類化合物(毛蕊異黃酮、毛蕊異黃酮苷、美的紫檀素、芒柄花苷、芒柄花素)的方法。黃芪樣品采用80%乙醇提取后,以超臨界CO20.2% H3PO4甲醇溶液為流動相,梯度洗脫,色譜柱為Waters ACQUITY UPC2 CSH柱(100 mm × 3.0 mm,1.8 μm),柱溫為40℃,流速為0.4 mL/min,進樣量為1 μL,檢測波長為280 nm。整個分析過程僅需15 min,分析速度是傳統液相色譜的3倍以上。結果表明:毛蕊異黃酮、毛蕊異黃酮苷、美的紫檀素、芒柄花苷與芒柄花素的檢出限及定量限范圍分別在0.3~0.5 mg/kg和1.0~2.0 mg/kg之間,5種黃酮類成分的加標平均回收率均高于99.7%,相對標準偏差(RSD)小于2.2%(n=6);在最優條件下,對13批不同產地的黃芪進行檢測,毛蕊異黃酮、毛蕊異黃酮苷、美的紫檀素、芒柄花苷與芒柄花素的含量范圍分別在4.8~102 mg/kg、14~277 mg/kg、0~135 mg/kg、5.3~119 mg/kg及2.8~41 mg/kg之間;本方法簡便快速,重復性良好,結果準確可靠,可用于黃芪藥材中5種主要黃酮類成分的含量測定。
關鍵詞 :超高效合相色譜; 黃酮類; 黃芪
1 引 言
超高效合相色譜法(Ultra performance convergence chromatography,UPC2)是超高效液相色譜(UPLC)和超臨界流體色譜(Supercritical fluid chromatography,SFC)技術的結合,它以超臨界流體二氧化碳為流動相主體,依靠流動相的溶劑化能力進行分離與分析[1,2]。相對于傳統超臨界流體色譜重復性較差的缺點,UPC2能夠通過精確調節流動相強度、壓力和溫度,在整個分離過程中獲得較好的重現性;此外,由于CO2超臨界流體具有較高的線速度及較低的密度,在分離過程中具有很高的柱效,且分析速度快,靈敏度高,有機溶劑使用量少。此外,CO2可與多數極性和非極性有機溶劑混溶,與氣相色譜(Gas chromatography,GC)相比,超臨界流體具有類似于液體的密度,為常壓氣體的200~500倍,具有較高的溶解能力,適于分離難揮發和熱不穩定性物質,而氣相色譜需將樣品氣化后才能進行分離操作,不適合于難揮發及熱不穩定物質的分離;與液相色譜(Liquid chromatography,LC)相比,超臨界流體具有較小的粘度,可減小過程阻力,在相同條件下,壓力降比液相色譜的低,并且具有較高的擴散系數和傳質速率,在分離操作時所用的時間短,單位時間內分離效能高[3~5]。
黃芪為常用補氣藥,《中華人民共和國藥典》2010年版一部載黃芪為豆料植物膜莢黃芪(Astragalus membranaceus (Fisch).Bge)或蒙古黃芪(A. membranaceus (Fisch )Bgevar.mongholicus (Bge) Hsiao)的干燥根,其味甘,性溫,歸肺、脾經;具有補氣固表、斂瘡生肌等作用,臨床用于氣虛乏力,表虛自汗,氣虛水腫,慢性腎炎蛋白尿和糖尿病等癥[6]。研究表明,黃酮類成分作為其有效成分之一,有抗菌抗病毒、降血脂、抗氧自由基等作用,還具有抗缺血和改善血象作用[7~9]。
近年對黃酮類成分含量測定方面的報道較多,常見的為高效液相色譜法[10~12],薄層色譜法[13~15],液相色譜串聯質譜法[16~19]等,對于液相色譜質譜法,因為儀器的價格昂貴,在國內還未被普遍使用;層析色譜及高效液相色譜法特異性不強,干擾物質較多、定量不準確,分析時間長,不能用于多種物質的同時測定。本研究采用超高效合相色譜法同時測定黃芪中5種主要黃酮類化合物,在15 min內實現了5種黃酮類化合物的分離。實驗表明,本方法快速準確、靈敏度高、重復性好、實用性強,為黃芪中5種主要黃酮類化合物的定性與定量檢測提供了一種高效可行的色譜檢測方法。
2 實驗部分
2.1 儀器、試劑與材料
超高效合相色譜儀(美國Waters公司),配有Waters EmpowerTM 3數據處理系統;3K30冷凍離心機(美國Sigma公司);MS3渦旋儀(德國IKA公司);移液槍(美國Thermo Electron公司,100~1000 μL、1.0~5.0 mL)。
毛蕊異黃酮、毛蕊異黃酮苷、美的紫檀素、芒柄花苷、芒柄花素(純度>98.5%,德國Dr. Ehrenstorfer GmbH公司);CO2(純度>99.997%,蘭州匯能公司);甲醇、乙腈、異丙醇、正己烷(色譜純,德國Merck KGaA公司);其余試劑均為分析純。
根據黃芪藥材的不同來源,將其依次編為1~13號,樣品均在產地購買,原植物由蘭州醫學院生藥植物室馬志剛副教授鑒定為多序巖黃芪Hedysarum polybolrys HandMazz,均為栽培品。
2.2 標準貯備液配制
稱取毛蕊異黃酮、毛蕊異黃酮苷、美的紫檀素、芒柄花苷和芒柄花素適量,用甲醇溶解并定容至100 mL,分別配制成含100 mg/L毛蕊異黃酮、130 mg/L毛蕊異黃酮苷、236 mg/L美的紫檀素、206 mg/L芒柄花苷和232 mg/L芒柄花素的標準儲備液,4℃下冷藏待用。
2.3 色譜條件
Waters Acquity UPC2 CSH Fluorophenyl色譜柱:(100 mm×3.0 mm, 1.8 μm);流動相:A為CO2, B為甲醇;流速0.4 mL/min;進樣量:1 μL;柱溫30℃;檢測波長280 nm;動態背壓(Active back pressure regulator,ABPR):11.72 MPa。梯度洗脫程序見表1。
2.4 樣品的制備
準確稱取粉碎后的黃芪樣品2.00 g于50 mL聚乙烯管中,加入30 mL 80%乙醇溶液,渦旋2 min混勻后,超聲提取30 min,重復提取2次,合并提取液,于80℃下減壓濃縮至近干,殘渣用乙醇定容至5 mL,用0.45 μm微孔濾膜過濾后,經超高效合相色譜分析,外標法定量。
3 結果與討論
3.1 色譜條件優化
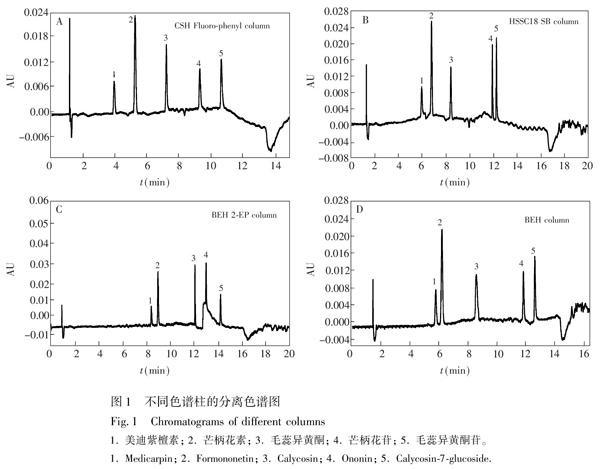
3.1.1 色譜柱的選擇
為了使黃芪中5種主要的黃酮類物質在較短分析時間達到分離并具有良好峰形,比較了Acquity UPC2 BEH 2EP(150 mm×2.1 mm i.d., 1.7 μm)、Acquity UPC2 HSS C18 SB(100 mm×3.0 mm i.d., 1.8 μm)、Acquity UPC2 BEH(150 mm×2.1 mm i.d., 1.7 μm)和Acquity UPC2 CSH Fluorophenyl(100 mm×3.0 mm i.d., 1.8 μm)這4款常見的色譜柱對5種主要黃酮類物質的分離影響(圖1)。由圖1可知,4種色譜柱均能很好地將5種黃酮類物質進行分離,但是使用BEH 2EP柱(圖1C)時,美迪紫檀素和芒柄花素出峰時間均較晚, 且芒柄花苷峰型不對稱,分離效果不理想;當使用HSS C18 SB柱和BEH柱時(圖1B和1D),雖然5種黃酮類物質均能達到分離要求,但是,對于HSS C18 SB柱,芒柄花苷和毛蕊異黃酮苷之間分離度較小,同樣,對于BEH柱,美迪紫檀素和芒柄花素之間分離度較小,在實際樣品檢測中,受到了未知物的干擾,影響定量分析準確性。而使用CSH Fluorophenyl柱時,5種黃酮類物質在12 min內出峰完全,且峰形尖銳,相互之間無影響。因此,本研究選擇Acquity UPC2 CSH Fluorophenyl色譜柱進行分離。
3.1.2 流速的選擇 由于超臨界CO2流體具有的較低的粘度和較高的擴散系數,使得超臨界流體作為流動相在分離過程中則具有較高的線速度;流動相的流速越大,被分離物質出峰時間越快,峰型尖銳;較小的流速會造成分析時間過長,色譜峰展寬,影響檢測的靈敏度。本實驗使用超高效合相色譜,對流速進行優化,考慮到助溶劑的加入和系統最高壓力的限制,流速在0.3~1.0 mL/min范圍內進行優化,當流速為0.3 mL/min時,分離時間長達18 min,不能做到快速、高通量分析,并且芒柄花苷和毛蕊異黃酮苷拖尾嚴重,影響定量分析的準確性及靈敏度。當流速大于0.6 mL/min時,由于梯度洗脫時間延長,助溶劑比例增加,系統壓力可能會超出最高壓力,為了保證較好的靈敏度、系統壓力以及目標物盡可能與雜質分離,本實驗的最佳流速選擇為0.4 mL/min。
3.1.3 助溶劑的選擇
由于UPC2的流動相主要是CO2超臨界流體,助溶劑的加入會使超臨界流體的溶劑化能力增強,從而使流動相對目標物的溶解性增大,能夠極大地改善目標化合物的峰形及保留時間,為了調整流動相的溶劑化能力,實驗中通常加入甲醇、乙醇、乙腈、異丙醇等助溶劑,有效改變目標化合物的峰形及保留時間。本實驗分別選用了甲醇、乙醇、異丙醇和乙腈4種常用的不同極性的助溶劑,對黃芪中5種主要黃酮類物質進行分離。結果表明,隨著助溶劑極性增大,流動相的溶解能力也相應增強,使得5種黃酮類物質出峰時間改變。在分別使用乙醇和異丙醇為助溶劑時,美迪紫檀素出峰時間提前,且由于乙醇和異丙醇的極性較小,芒柄花苷和毛蕊異黃酮苷在流動相中的溶解能力降低,出現較為嚴重的色譜峰拖尾;使用乙腈作為助溶劑時,由于流動相具有較低的溶劑化能力,使得芒柄花苷和毛蕊異黃酮苷色譜峰拖尾嚴重。當選用甲醇為助溶劑時,5種黃酮類物質在較短時間內達到較好分離,并具有較好的峰型,因此,本實驗選擇甲醇為助溶劑。
3.1.4 動態背壓(ABPR)的選擇
超高效合相色譜中,動態背壓(ABPR)控制CO2在整個操作過程中的超臨界流體狀態, 是影響分離過程的重要因素之一。不同的動態背壓下,CO2超臨界流體對各種樣品有著不同的溶解能力。當背壓升高時,超臨界流體密度增大,溶劑化能力增強,柱壓升高,分析物保留時間提前。本實驗在10.31~14.48 MPa范圍內考察了CO2超臨界流體對樣品分離度的影響。結果表明,隨著背壓增大,CO2超臨界流體密度及黏度隨之增加,柱壓升高;當背壓為10.31 MPa時,5種黃酮類物質雖然得到較好分離,但是分析時間較長,達到了17 min;當背壓高于13.1 MPa時,系統壓力已超過最高壓力的80%。綜合考慮動態背壓及系統壓力,當背壓為11.72 MPa時,5種黃酮類物質之間分離情況最好,保留時間適中、峰形對稱,故本實驗選擇動態背壓為11.72 MPa。
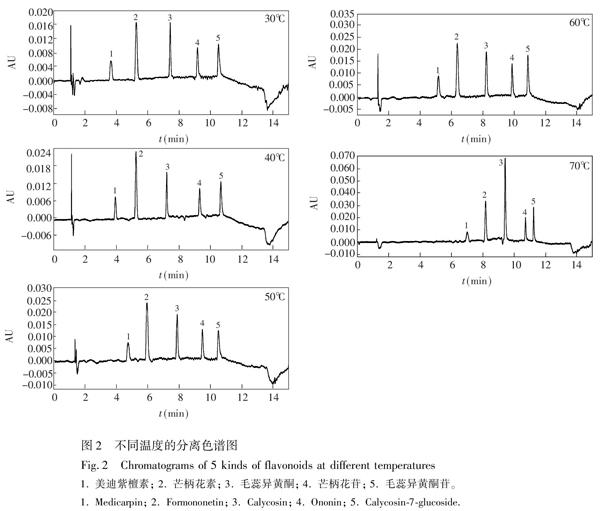
3.1.5 色譜柱溫度的選擇
隨著溫度升高,超臨界流體的黏度降低,溶劑化能力減小,使得目標物出峰時間延長;隨著溫度降低,超臨界流體黏度增加,溶劑化能力增強,目標物出峰時間相應的縮短,這與傳統的液相色譜相反。為了使樣品中5種黃酮類物質得到較好分離,本實驗在保持其它色譜條件不變的前提下,在30℃~70℃范圍內考察了柱溫對目標物分離的影響(圖2)。結果表明,隨著溫度升高,黃酮類物質的保留時間逐漸延長,當溫度為40℃時,目標物均得到分離; 當溫度逐漸升高時,目標物的出峰時間延長,分離度減小,其中所含雜質易干擾目標物。因此,本實驗選擇最佳分離溫度為40℃。
3.1.6 提取溶劑及提取條件的選擇
為了盡可能多且完全地提取黃芪中的有效成分,本實驗分別以50%~90%甲醇以及50%~90%乙醇對黃芪藥材進行超聲提取。結果表明,80%乙醇超聲提取,黃芪提取液中5種主要黃酮類物質的相對含量最多。本研究同樣考察了提取時間和提取次數對提取效率(峰面積)的影響。結果表明,30 mL 80%乙醇提取兩次,每次30 min,提取效率最高。
3.2 方法學考察
3.2.1 穩定性實驗 取黃芪樣品,按照2.4節處理樣品,分別在2.3節所述色譜條件下進樣分析 0, 4, 12, 24和48 h,黃芪中的美迪紫檀素、芒柄花素、毛蕊異黃酮、芒柄花苷、毛蕊異黃酮苷的峰面積RSD分別為0%, 0.5%, 0.5%, 0.7%和0.8%。表明樣品溶液在48 h內穩定。
3.2.2 專屬性實驗 取所得1號樣品溶液、混合標準溶液、陰性供試品溶液,在2.3節所述的色譜條件下進樣分析,將陰性樣品與混合標準溶液色譜圖和樣品色譜圖比較(圖3),在5種黃酮類的出峰位置無干擾,表明本方法專屬性好。
3.2.3 線性范圍及定量限
分別量取5種黃酮類物質的對照品溶液1.0, 2.5, 5.0, 10.0, 20.0和50.0 mL,置于100 mL棕色容量瓶中,用甲醇溶解并定容,得到0.10~118.0 mg/g的系列溶液。在2.3節所述色譜條件下,每個濃度依次進樣1.0 μL,以濃度(μg/mL)為橫坐標,以峰面積(A)為縱坐標,進行線性回歸。由表2可見,5種黃酮類物質在一定范圍內線性關系良好。
3.2.4 精密度實驗
按照2.3節所述色譜條件,5種黃酮類物質標準溶液重復進樣6次,日內精密度(RSD)在0.5%~0.8%之間;日間精密度(RSD)在0.5%~1.1%之間,可以看出本方法對檢測黃芪中5種黃酮類物質具有較好的日間和日內精密度,可滿足黃芪中5種主要黃酮類物質的檢測要求。
3.2.5 重復性實驗
準確稱取同一黃芪樣品(No.1)6份,按2.4節進行樣品處理,在2.3節所述的色譜條件下進樣分析,樣品色譜圖見圖3, 5種黃酮類化合物的含量及相對標準偏差(RSD)計算結果見表3,同一份黃芪樣品(No.1)中美迪紫檀素、芒柄花素、毛蕊異黃酮、芒柄花苷、毛蕊異黃酮苷含量的RSD分別為1.3%, 2.6%, 2.7%, 2.7%, 2.2%。表明本方法重復性較好。
3.2.6 加標回收率 準確稱取6份同一批(No.1)黃芪樣品,準確加入5種被測組分混合標準品的乙醇溶液1 mL,按按2.4節進行樣品處理,在2.3節所述的色譜條件下進樣分析(表4),5種被測組分加樣回收率在98.8%~103.5%之間,RSD在1.4%~2.1%之間,表明本方法準確可靠。
3.3 實際樣品分析
取所收集的13批次黃芪樣品(粉碎后過4號篩),準確稱取2.00 g,按2.4節進行樣品處理,在2.3節所述的色譜條件進樣分析(表5)。部分黃芪樣品色譜圖見圖4。
3.4 UPC2與HPLC對比分析
按2.5節進行樣品處理,用Waters Alliance 2695高效液相色譜儀進樣10 μL進行分析,流動相為甲醇0.2% H3PO4溶液,梯度洗脫,280 nm下檢測。另取相同供試品溶液,用Waters 超高效合相色譜進樣分析,進樣量為1 μL,流動相為甲醇0.2% H3PO4溶液,梯度洗脫,280 nm下檢測。分別得到1號黃芪樣品的超高效合相色譜和高效液相色譜的色譜圖,分別見圖3及圖5。由圖5可見,超高效合相色譜法與高效液相色譜法相比,因為保留機理不同,5種黃酮類化合物在超高效合相色譜與高效液相色譜的分離過程中,出峰順序完全相反,且高效液相色譜分析時間長達90 min,而超高效合相色譜分析時間較高效液相色譜縮短近4倍。由此可見,傳統液相色譜方法相比,本實驗建立的超高效合相色譜快速檢測黃芪中5種主要黃酮類化合物方法即節省溶劑,降低檢測成本,又能使分析物快速分離,實現高通量檢測。
4 結 論
采用超高效合相色譜法同時測定了黃芪中的5種主要的黃酮類成分。本方法靈敏度高,重現性、穩定性良好,分析時間短,除平衡儀器所用的5 min外,完成一次分析僅15 min,分析速度比高效液相色譜快近4倍,可用于黃芪藥材中黃酮類成分含量的高通量測定。采用本方法對13批不同來源的黃芪藥材的5種主要黃酮類物質的含量進行測定,結果含量差異較大,這可能是由于藥材的品種、產地、種植方式及氣候等因素導致。
References
1 ZHOU Wei, WANG Bo, LIUQianQian, YANG ShengXin, WANG LiTing. Chinese J. Anal. Chem., 2015, 43(1): 115-120
周 圍, 王 波, 劉倩倩, 楊盛鑫, 王麗婷. 分析化學, 2015, 43(1): 115-120
2 WANG Bo, YAN Heng, WANG SuJun,LIU AJing, YANG ShengXin, ZHOU Wei. Journal of Instrumental Analysis, 2015, 34(7): 824-828
王 波, 閆 衡, 王肅軍, 劉阿靜, 楊盛鑫, 周 圍. 分析測試學報, 2015, 34(7): 824-828
3 XU YongWei, SUN QingLong, HUANG Jing, TAN XiaoJie. Modern Instruments, 2012, 18(5): 45-48
徐永威, 孫慶龍, 黃 靜, 譚曉杰. 現代儀器, 2012, 18(5): 45-48
4 GrandGuillaume Perrenoud A, Veuthey J L, Guillarme D. J. Chromatogr. A, 2012, 1266: 158-167
5 WANG LiTing, WANG Bo, ZHOU Wei, LIU QianQian, YANG ShengXin, ZHANG YaHeng. Chinese J. Anal. Chem., 2015, 43(7): 1047-1052
王麗婷, 王 波, 周 圍, 劉倩倩, 楊盛鑫, 張雅珩. 分析化學, 2015, 43(7): 1047-1052
6 National Pharmacopoeia Committee. Pharmacopoeia of the People′s Republic of China (Part Ⅰ). Beijing: China Medical Science and Technology Press, 2010: 284
國家藥典委員會.中華人民共和國藥典(一部). 北京:中國醫藥科技出版社, 2010: 284
7 HU FangDi, FENG ShiLan, ZHAO JianXiong. Journal of Chinese Medicinal Materials, 2003, 26(9): 634
胡芳弟, 封士蘭, 趙健雄. 中藥材, 2003, 26(9): 634
8 QIU YongBo, LIU Jin, WU Fei. Chinese Journal of Convalescent Medicine, 2011, 5: 435-436
邱勇波, 劉 錦, 武 飛. 中國療養醫學, 2011, 5: 435-436
9 CHEN JianZhen, L GuiYuan, YE Lei, CHEN JianMing. Herald of Medicine, 2009, 28(10): 1314-1316
陳建真, 呂圭源, 葉 磊, 陳建明. 醫藥導報, 2009, 28(10): 1314-1316
10 LIANG Jin, FENG ShiLan, LIU XiaoHua, DANG ZiLong, LIANG JianDi, YANG ChunXia. Northwest Pharmaceutical Journal, 2012, 27(5): 490-493
梁 瑾, 封士蘭, 劉小花, 黨子龍, 梁建娣, 楊春霞. 西北藥學雜志, 2012, 27(5): 490-493
11 LUO ChunXia, LIN PingChuan, GU LiHua, WU Tao, WU DaZheng, WANG ZhengTao, HU ZhiBi. China Journal of Chinese Materia Medica, 2003, 28(7): 603-605
羅春霞, 林平川, 谷麗華, 吳 弢, 吳大正, 王崢濤, 胡之璧. 中國中藥雜志, 2003, 28(7): 603-605
12 WANG XiaoHui, LIU Tao, LI Qing, CHEN XiaoHui, BI KaiShun. Chinese Journal of Chromatography, 2006, 24(6): 486-488
王曉輝, 劉 濤, 李 清, 陳曉輝, 畢開順. 色譜, 2006, 24(6): 486-488
13 LIU XiaoQing, LI Jun, HU JingLian, LI XiaoYan, MEN QiMing, KANG TingGuo . Journal of Liaoning University of Traditional Chinese Medicine, 2013, 15(07): 53-55
劉曉慶, 李 軍, 胡景蓮, 李曉艷, 門啟鳴, 康廷國. 遼寧中醫藥大學學報, . 2013, 15(07): 53-55
14 LIANG CongQing, ZHOU Xuan, SONG FenYun, ZHONG ZhaoJian. Chinese Journal of Experimental Traditional Medical Formulae, 2004, 10(5): 9-11
梁從慶, 周 漩, 宋粉云, 鐘兆健. 中國實驗方劑學雜志, 2004, 10(5): 9-11
15 LIANG XuMing, TANG YaoShu, ZHANG XiaoMei. LiShiZhen Medicine and Materia Medica Research, 2007, 18(1): 22-25
梁旭明, 唐耀書, 張小梅. 時珍國醫國藥, 2007, 18(1): 22-25
16 WU DingHui, CHEN GuanJian. Journal of Practical Traditional Chinese Medicine, 2012, 28(1): 65-67
吳定慧, 陳關鍵. 實用中醫藥雜志, 2012, 28(1): 65-67
17 ZHANG JiaYu, QIAO YanJiang, ZHANG Qian, GAO XiaoYan, LU JianQiu. Chinese Journal of Pharmaceutical Analysis, 2013, 27(2): 349-354
張加余, 喬延江, 張 倩, 高曉燕, 盧建秋. 藥物分析雜志, 2013, 27(2): 349-354
18 TANG GuiBao, CHEN Nan, PAN Xin. Strait Pharmaceutical Journal, 2013, 23(12): 7-11
唐瑰寶, 陳 楠, 潘 馨. 海峽藥學, 2013, 23(12): 7-11
19 CHEN Ting, TIAN Feng, TANG YueNian, LIU Yan, LIN ZhiYan, WANG YeChen. China Pharmacist, 2014, 17(4): 593-596
陳 婷, 田 豐, 唐躍年, 劉 艷, 林志燕, 王曄塵. 中國藥師, 2014, 17(4): 593-596
Abstract A method was developed for the rapid separation and determination of 5 kinds flavonoids (calycosin, calycosin7OβDglucoside, ononin, medicarpin and formononetin) in Astragali Radix based on ultraperformance convergence chromatography. After extracting by 80% ethanol, the flavonoids were separated on a Waters Acquity UPC2 CSH (100 mm×3.0 mm i.d., 1.8 μm) column at 40℃ by using supercritical CO2methanol (contain 0.2% H3PO4) acetonitrile as the mobile phase at a flow rate of 0.4 mL/min, and then analyzed by a UV detector at wavelength of 280 nm, the whole analysis progress was only 15 min. The results showed that the limits of detection (LOD) and the limits of quantitation (LOQ) of five flavonoids were between 0.3 and 0.5 mg/kg, and 1.0 and 2.0 mg/kg, respectively. The spiked recoveries were more than 99.7%, and the relative standard deviations (RSD) were less than 2.2% (n=6). Under the optimal conditions, 13 groups of Astragali Radix from different producing areas were detected. The contents of calycosin, calycosin7OβDglucoside, medicarpin, ononin and formononetin were 4.8-102 mg/kg, 14-277 mg/kg, 0-135 mg/kg, and 5.3-119 mg/kg, 2.8-41 mg/kg, respectively. This method is simple, fast, accurate and reproducible, and the result is reliable. The method is applicable for the determination of 5 flavonoids in Astragali Radix.
Keywords Ultraperformance convergence chromatography; Flavonoids; Astragali Radix
