雙亞基丹酚酸酯酶基因的表達載體構建及其生物轉化
2016-09-13 01:20:16劉春瑩李泰厚魚紅閃
食品工業科技 2016年13期
程 妍,劉春瑩,李泰厚,魚紅閃,*
(1.大連工業大學 生物工程學院,遼寧大連 116034;2.韓國慶熙大學 生命科學學院漢方藥材料與加工,韓國京畿道龍仁市 446-701)
?
雙亞基丹酚酸酯酶基因的表達載體構建及其生物轉化
程妍1,劉春瑩1,李泰厚2,魚紅閃1,*
(1.大連工業大學 生物工程學院,遼寧大連 116034;2.韓國慶熙大學 生命科學學院漢方藥材料與加工,韓國京畿道龍仁市 446-701)
為了實現丹酚酸酯酶的外源性表達,利用了大腸桿菌和畢赤酵母兩種表達系統,原核大腸桿菌采用pET 28a和pET 32a構建表達載體,并對重組大腸桿菌進行分別誘導表達和共表達;真核畢赤酵母采用pPIC9K構建共表達載體,對重組畢赤酵母進行誘導表達。研究結果表明,兩種體系均能誘導表達出丹酚酸酯酶,蛋白在大腸桿菌系統中得到了高效表達,但未顯示出該酶活力。畢赤酵母系統可使蛋白分泌性表達,表達產物具有一定的丹酚酸酯酶活力,但表達量不高。說明大腸桿菌系統體系更為適合大量表達丹酚酸酯酶,由此提供了一種該酶的外源表達方法。
丹酚酸酯酶,大腸桿菌,畢赤酵母,克隆表達
丹參為唇形科鼠尾草屬植物丹參(Salvia miltiorrhiza Bunge)的干燥根及根莖,具有祛瘀通絡,清心安神等功效[1]。近年來對丹參的藥理活性研究表明,丹參中的丹參素,又名D(+)β-(3,4-二羥基苯基)乳酸,具有很強的抗脂質過氧化和清除自由基作用,是降低心肌梗死、抗動脈粥樣硬化、抑制血栓形成的有效成分,在心腦血管系統疾病防治等方面應用廣泛[2]。但天然丹參中丹參素的含量比較低(約0.5%),且丹參植物資源有限,這大大限制了丹參素類制劑的生產及研發。
丹酚酸B在丹參中的含量比較高(約2%~8%),而且丹酚酸B是由一分子咖啡酸和三分子丹參素酸縮合而成的化合物,內含酯鍵。目前已有采用超高壓提取[3]、微波法[4]、堿法[5]提取丹參中丹參素的文獻報道,但關于生物酶法轉化丹酚酸B生成丹參素的研究卻寥寥無幾。本實驗室已篩選到一株Absidiasp. D30s菌,所產酶能定向水解丹酚酸B上的酯鍵生成丹參素,暫命名為丹酚酸酯酶[6-7],分離純化后發現該酶由分子質量分別為38 ku(亞基Ⅰ)和18 ku(亞基Ⅱ)的兩個亞基組成,并調取了丹酚酸酯酶2個亞基的基因全序列。Blast分析顯示,亞基Ⅰ序列與堿性蛋白酶基因序列、亞基Ⅱ序列與Cu,Zn超氧化物歧化酶基因序列,相似性很高,但這些酶都與水解丹酚酸B上的酯鍵毫無關系,表明新篩選到的丹酚酸酯酶是一種特異性的酶蛋白。

表1 克隆引物
本研究克隆并構建了兩種應用于丹酚酸酯酶的表達載體,對該酶基因在大腸桿菌和畢赤酵母中的異源表達進行了初步研究,以期尋求較為適合大量表達該酶的表達系統。一方面進一步驗證調取基因的正確性,另一方面,為構建生物合成丹酚酸酯酶的工程菌及其規模化制備提供參考。
1 材料與方法
1.1材料與儀器
E.cloiBL21(DE3)、pET 28a(卡那霉素抗性)和pET 32a(氨芐青霉素抗性)寶生物工程(大連)有限公司;畢赤酵母和pPIC9K載體由韓國慶熙大學李泰厚教授實驗室提供,共轉化雙亞基基因的畢赤酵母是在韓國慶熙大學李泰厚教授實驗室完成;限制性核酸內切酶、低分子質量蛋白Marker、PrimeSTAR?HS DNA Polymerase、TaKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver.3.0、In-Fusion?HD Cloning Kit寶生物工程(大連)有限公司;實驗所用試劑均為國產分析純。
BioSafer9700 PCR熱循環儀賽飛生物科技有限公司;UV-120-02型分光光度計日本京都Shimadzu公司;HimacCF15R型高速冷凍離心機賽默飛世爾科技公司;JM-250型電泳儀大連競邁生物科技有限公司。
1.2實驗方法
1.2.1目的基因擴增以實驗室前期反轉錄得到的雙亞基丹酚酸酯酶cDNA序列為模板,設計克隆引物(表1)。使用PrimeSTAR?HS DNA Polymerase PCR擴增亞基Ⅰ基因CDS區1212 bp,片段兩側添加NcoⅠ和XhoⅠ酶切位點,擴增亞基Ⅱ基因CDS區465 bp,片段兩側添加BalⅠ和XhoⅠ酶切位點。PCR條件:98 ℃變性2 min,98 ℃變性10 s,55 ℃退火10 s,72 ℃延伸60 s,完成30個循環后72 ℃延伸10 min。將PCR產物加A連接至pGM-T載體后委托寶生物(大連)有限公司進行測序。
1.2.2重組質粒(pET 28a和pET 32a)構建與大腸桿菌轉化將載體pGMT-Ⅰ(亞基Ⅰ)和表達載體pET 28a分別使用NcoⅠ/XhoⅠ進行酶切,載體pGMT-Ⅱ(亞基Ⅱ)和表達載體pET 32a分別使用BalⅠ/XhoⅠ進行酶切。酶切產物連接,熱轉化至E.coliCompetent Cell JM109中,涂布平板,37 ℃過夜培養。挑選陽性菌落植菌,提取質粒,委托寶生物(大連)有限公司進行測序。提取重組質粒pET 28a-Ⅰ(亞基Ⅰ)和pET 32a-Ⅱ(亞基Ⅱ)分別熱轉化至大腸桿菌BL21(DE3)中。將等量pET 28a-Ⅰ和pET 32a-Ⅱ質粒共轉化大腸桿菌BL21(DE3)中,涂布抗性平板篩選單菌落。
1.2.3重組質粒在大腸桿菌中誘導表達含pET 28a-Ⅰ重組菌接入含卡那霉素的LB培養基中,含pET 32a-Ⅱ重組菌接入含氨芐抗生素LB培養中,共含pET 28a-Ⅰ和pET 32a-Ⅱ重組菌接入含卡那霉素和氨芐抗生素雙抗性的LB培養中,37 ℃振蕩培養過夜,2%接種量接種至新的含相應抗生素的LB培養基中,37 ℃振蕩培養至OD600為0.6~0.8,加入IPTG(終濃度0.5 mmol/L),37 ℃誘導表達4 h后離心收集菌體,用PBS(pH7.4)緩沖液重懸洗滌兩次,加入PBS重懸,超聲破碎離心收集上清液和沉淀,采用硫銨沉淀及透析技術[8]對上清液進行濃縮(可用于酶活性檢測)并進行SDS-PAGE(5%濃縮膠,12%分離膠)檢測[9]。
1.2.4重組質粒在畢赤酵母中誘導表達將整合入丹酚酸酯酶雙亞基基因的畢赤酵母接種到25 mL BMGY培養基中,28 ℃下振蕩培養18 h,至OD600達到2.0~6.0。離心收集菌體重懸于25 mL BMMY培養基中,28 ℃振蕩培養144 h,每隔24 h補加100%甲醇(終濃度0.5%)。將培養液離心,收集發酵液上清,采用硫銨沉淀及透析技術對上清液進行濃縮(可用于酶活性檢測)并進行SDS-PAGE(5%濃縮膠,12%分離膠)檢測前可再經TCA濃縮。
1.2.5表達產物酶活性的TLC檢測上述大腸桿菌和畢赤酵母的重組菌經誘導培養,發酵液濃縮與0.1%的丹酚酸B溶液按體積比1∶1混勻,于40 ℃反應16 h,加入飽和正丁醇終止反應,展開劑為乙酸乙酯∶甲酸=10∶1,均勻噴灑顯色劑(5% FeCl3),利用TLC法[10]檢測酶活性。
2 結果與分析
2.1亞基Ⅰ和亞基Ⅱ基因的克隆
以雙亞基丹酚酸酯酶cDNA序列為模板,PCR擴增,得到了大小約為1200 bp和500 bp的DNA片段(圖1),與理論值(亞基Ⅰ 1212 bp,亞基Ⅱ 465 bp)相符。將PCR產物加A連接至pGM-T載體后送至寶生物(大連)有限公司進行測序。測序顯示,亞基Ⅰ基因CDS區長1212 bp;亞基Ⅱ基因CDS區長465 bp,與參考序列一致。
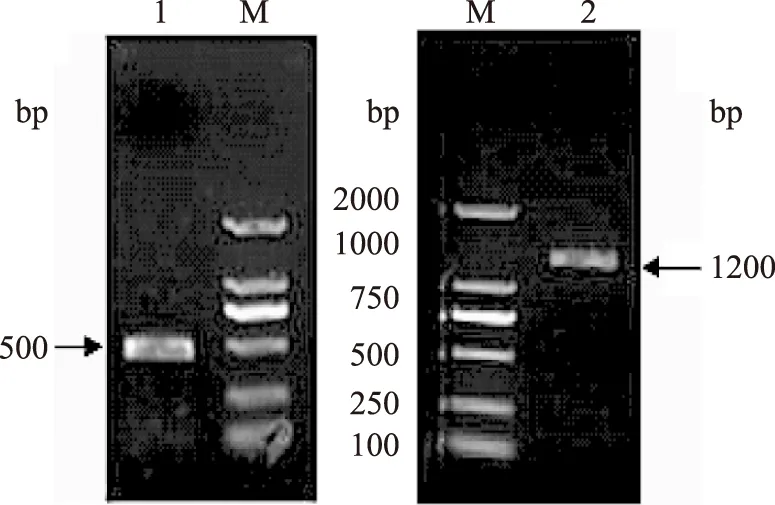
圖1 PCR擴增目的基因Fig.1 Amplification of insert DNA by PCRM:DL 2000 DNA Marker;注:1:亞基Ⅱ PCR擴增;2:亞基Ⅰ PCR擴增。
2.2重組質粒pET28a和pET32a的構建
經瓊脂糖凝膠電泳可見,pGMT-Ⅰ經NcoⅠ/XhoⅠ酶切后得到亞基Ⅰ產物大小約為1212 bp,由于上樣濃度高,而出現了pGMT-Ⅰ酶切不完全現象(圖2a泳道1);載體pGMT-Ⅱ經BalⅠ/XhoⅠ酶切后得到亞基Ⅱ大小約為465 pb(圖2a泳道2);空載體pET 28a經NcoⅠ/XhoⅠ酶切后得到產物大小約為5400 bp(圖2b泳道3);空載體pET 32a經BalⅠ/XhoⅠ酶切后得到產物大小約為5900 bp(圖2b泳道4),均與理論值相符,回收酶切產物。按1.2.2方法得到重組質粒pET 28a-Ⅰ和pET 32a-Ⅱ,送至寶生物(大連)有限公司進行測序。測序結果與參考序列一致,表明表達質粒pET 28a-Ⅰ和pET 32a-Ⅱ構建成功。
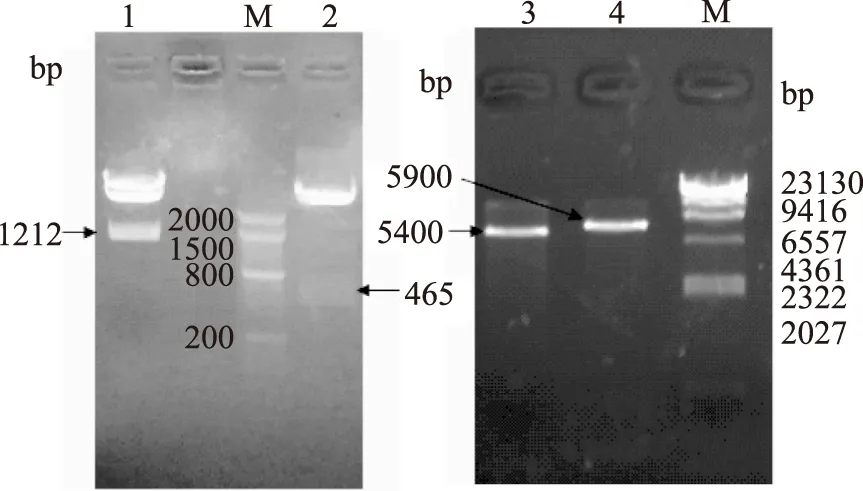
圖2 pGMT-Ⅰ、pGMT-Ⅱ、pET 28a、pET 32a酶切產物回收Fig.2 Enzyme digestion of pGMT-Ⅰ、pGMT-Ⅱ、pET 28a、pET 32a注:a:pGMT-Ⅰ、pGMT-Ⅱ酶切產物;b:空載體pET 28a、空載體pET 32a酶切產物;M:Marker;a1:pGMT-Ⅱ經BalⅠ/XhoⅠ酶切;a2:pGMT-Ⅰ經NcoⅠ/XhoⅠ酶切;b3:pET 28a經NcoⅠ/XhoⅠ酶切;b4:pET 32a經BalⅠ/XhoⅠ酶切。
2.3重組質粒在大腸桿菌中的誘導表達
按1.2.3方法得到外源目的基因單獨誘導表達產物,經SDS-PAGE檢測(圖3、圖4),分別與空質粒pET 28a和空質粒pET 32a轉化大腸桿菌BL21(DE3)作對照,結果顯示均有一條明顯的蛋白表達差異條帶,并且表達量占菌體總蛋白量較高,表明外源目的基因均能高效表達。但蛋白質表觀分子質量都較核酸序列推測所得的理論分子質量(38 ku和18 ku)偏大。
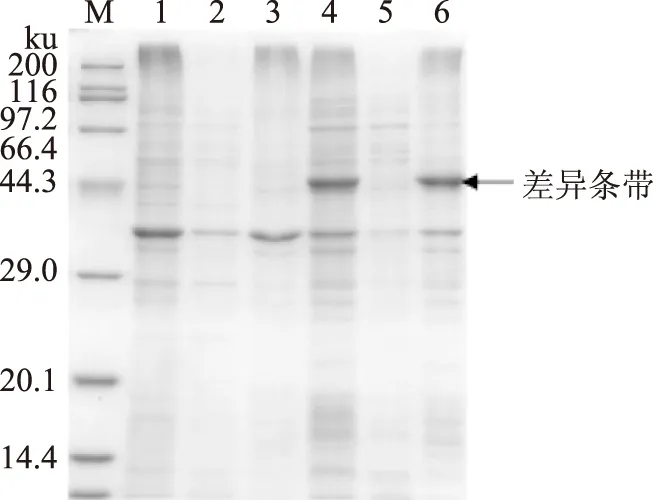
圖3 SDS-PAGE電泳分析(亞基Ⅰ)Fig.3 SDS-PAGE analysis(subunit Ⅰ)注:M,Protein MW marker;1,pET 28a全細胞;2,pET 28a上清;3,pET 28a沉淀;4,破碎全細胞;5,破碎上清;6,破碎沉淀。
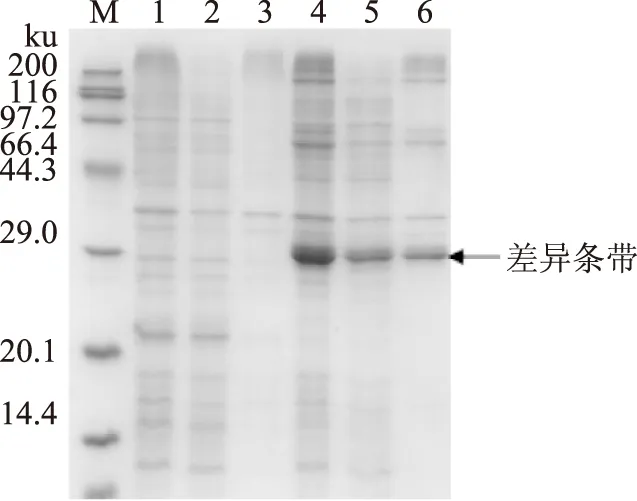
圖4 SDS-PAGE電泳分析(亞基Ⅱ)Fig.4 SDS-PAGE analysis(subunit Ⅱ)注:M,Protein MW marker;1,pET 32a全細胞;2,pET 32a上清;3,pET 32a沉淀;4,破碎全細胞;5,破碎上清;6,破碎沉淀。
pET 28a-Ⅰ、pET 32a-Ⅱ分別轉化大腸桿菌BL21(DE3)后,經誘導都可高效表達目的蛋白,且目的蛋白占菌體總蛋白量較高,但存在表觀分子質量與理論分子質量不一致的現象。Simone[11]等人和Richards[12]等人的研究中都已報道過表觀分子質量與預測不一致的現象,但產生偏差的原因還尚未完全闡明,推測可能與不合適的電泳條件[13]、結構的變化或電荷分布不均[14]等有關,侯穎[15]等利用大腸桿菌系統表達得到過表觀分子質量偏高于理論值的重組蛋白,分析后確定為短肽間單獨的巰基與谷胱甘肽發生了縮合反應所引起的。由于導致分子質量偏差的因素較多,更具體的原因還有待于今后進一步的研究。
按1.2.3方法得到外源目的基因共表達產物,與共轉入空質粒pET 28a和空質粒pET 32a的大腸桿菌BL21(DE3)對照,在位置分別與pET 28a-Ⅰ和pET 32a-Ⅱ單獨轉化大腸桿菌BL21(DE3)所表達的重組蛋白質相同處,有兩條新增蛋白質帶,結果顯示重組質粒中的目的基因在陽性克隆誘導系統中有表達,但重組質粒pET 32a表達不明顯(圖5)。
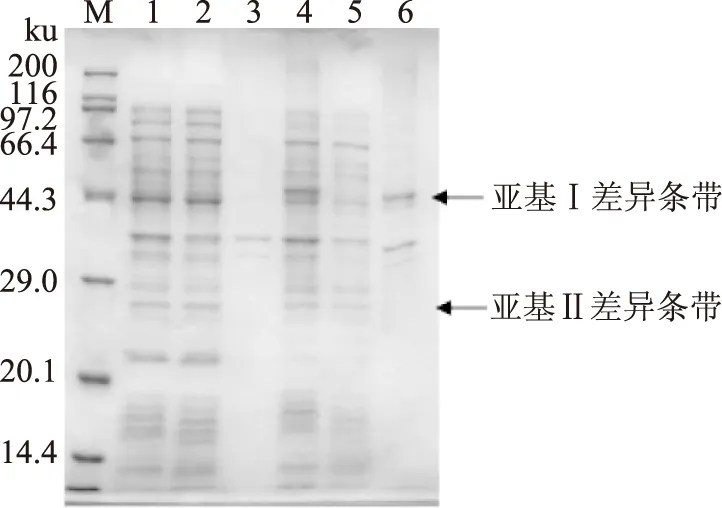
圖5 SDS-PAGE電泳分析(亞基Ⅰ+亞基Ⅱ)Fig.5 SDS-PAGE analysis(subunit Ⅰ+subunit Ⅱ)注:M,Protein MW marker;1,pET 28a+pET 32a全細胞;2,pET 28a+pET 32a上清;3,pET 28a+pET 32a沉淀;4,破碎全細胞;5,破碎上清;6,破碎沉淀。
一般認為,復制子相同的兩種質粒不能同時穩定地保持在一個細胞內,兩種質粒在復制及分配到子代細胞過程中彼此競爭,容易導致子代細胞中兩種質粒復制的失衡,而易丟失一種質粒[16],質粒pET 28a和質粒pET 32a具有相同復制子,外源基因單獨表達時表達量較高,在被共同轉入一個細胞時,推測可能因為質粒的不相容性而發生了質粒復制的不均等現象,從而形成了蛋白質表達量的不一致,但仍實現了丹酚酸酯酶亞基Ⅰ和亞基Ⅱ在大腸桿菌中的共表達。
2.4重組質粒在畢赤酵母中的誘導表達
丹酚酸酯酶基因在畢赤酵母中共表達方面的研究,與韓國慶熙大學李泰厚教授團隊共同合作完成,本實驗室前期調取的亞基Ⅰ及亞基Ⅱ基因在慶熙大學進行了pPIC9K共表達載體的構建。以pGMT-Ⅰ為模板,PCR擴增亞基Ⅰ基因CDS區1212 bp,片段兩側添加EcoRⅠ和NotⅠ酶切位點,克隆至pPIC9K表達載體中,PmeI線性化后電轉化至畢赤酵母中。以pGMT-Ⅱ為模板,PCR擴增亞基Ⅱ基因CDS區465 bp,片段兩側添加EcoRⅠ和NotⅠ酶切位點,克隆至pPIC9K表達載體中,SacI線性化后電轉化至上述已整合上亞基Ⅰ基因的畢赤酵母中,測序檢測亞基Ⅰ基因和亞基Ⅱ基因,實現共轉化。
重組畢赤酵母經誘導培養所得的發酵液上清進行SDS-PAGE測定(圖6),經與未添加甲醇誘導的畢赤酵母轉化子做對照,在約38 ku和約18 ku處各得到一條蛋白表達差異條帶,與核酸序列推測所得的理論分子質量基本不一致,表明重組質粒pPIC9K在畢赤酵母中表達成功,丹酚酸酯酶實現了在畢赤酵母中的表達,但表達量不高。
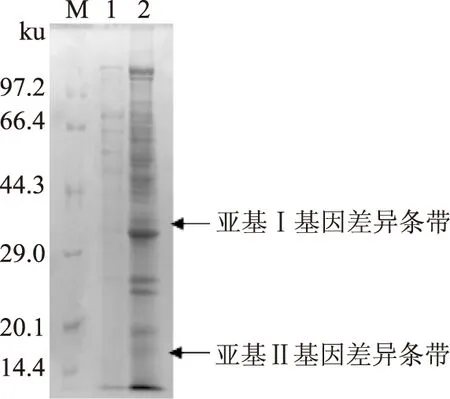
圖6 SDS-PAGE電泳分析(亞基Ⅰ+亞基Ⅱ)Fig.6 SDS-PAGE analysis(subunit Ⅰ+subunit Ⅱ)注:M,Protein marker;1,無甲醇誘導菌株;2,甲醇誘導菌株。
2.5表達產物TLC活性檢測
重組質粒pET 28a-Ⅰ和pET 32a-Ⅱ的共轉化大腸桿菌表達產物經酶反應后進行TLC活性檢測結果(圖7)顯示,沒有酶反應產物產生,說明其產物未具有丹酚酸酯酶活性。畢赤酵母表達產物經酶反應后進行TLC活性檢測,得到了酶反應產物,說明其表達產物具有一定的丹酚酸酯酶的催化活性。
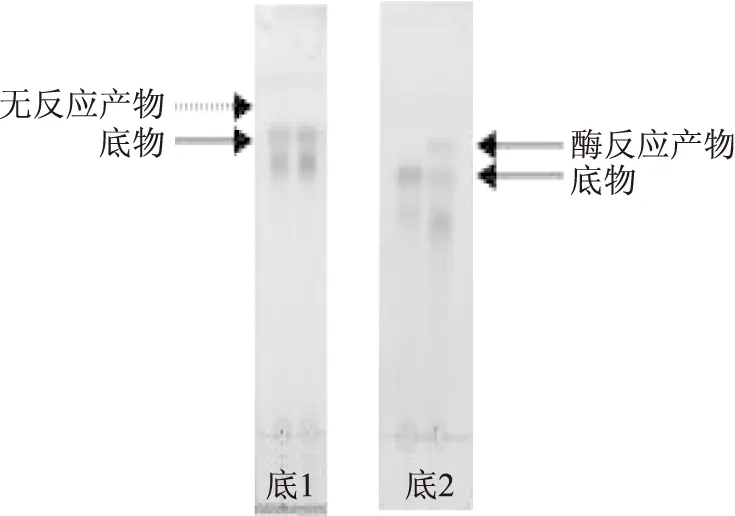
圖7 大腸桿菌表達產物及畢赤酵母表達產物酶反應薄層層析圖Fig.7 TLC of E. coli expression products and Pichia pasioris expression products注:底,0.1%丹酚酸B溶液和水反應;1,共表達大腸桿菌表達產物酶反應產物;2,畢赤酵母表達產物酶反應產物。
如2.3所述,大腸桿菌中外源基因各自單獨表達時,均可高效表達目的蛋白質(圖3、圖4),關于丹酚酸酯酶在大腸桿菌中能夠高效表達的原因目前并不十分清楚,推測這可能與表達系統特性和外源基因本身特性這兩方面有關。Grosjean[17]等的研究顯示,在翻譯的過程中,宿主菌存在著對密碼子的偏好性,丹酚酸酯酶在大腸桿菌中能夠高效表達,一種可能原因是基因序列中高頻出現的密碼子在大腸桿菌中很多見。
大腸桿菌表達的重組蛋白質未顯示出丹酚酸酯酶活力,推測有如下幾種可能原因。重組蛋白在大腸桿菌中高效表達時,可能由于不能正確折疊而形成了無活性的包涵體,李單單[18]等的文獻報道,外源基因在表達時包涵體的出現可能也會影響其具有生物活性的四級結構的形成。外源基因共表達時,兩種蛋白質的表達量存在差異(圖5),這也可能會影響到雙亞基蛋白質分子的高級結構組裝,進而影響其酶活力的發揮。Large[19]等的文獻報道,蛋白質分子量的偏差變化可能會對蛋白質的組裝產生影響,從而影響其生物活性的發揮。大腸桿菌為原核表達系統,其也可能因缺少真核生物的翻譯后修飾系統而使蛋白表達不出活性[20]。丹酚酸酯酶在大腸桿菌中高效表達的外源蛋白包涵體富含重組蛋白,進行有效的變復性處理后,獲得大量得丹酚酸酯酶將成為可能。目前,應用最普遍的復性方法有稀釋、透析、超濾等,其中稀釋法因其簡單、有效而成為許多重組蛋白包涵體復性的首選方法[21],近年來,色譜技術也廣泛的應用到了蛋白質復性中[22],但由于丹酚酸酯酶相對分子質量偏差等因素的影響,該酶的變復性條件仍需要進行進一步探討。
畢赤酵母表達系統由于具備對翻譯后蛋白質進行修飾和加工的生物環境,使得分泌表達出的丹酚酸酯酶具有一定的生物學活性(圖7),但表達量并不高(圖6)。雖然許多蛋白質在畢赤酵母表達系統中的表達量很高,但也有些蛋白質的表達量很低,Fidler[23]等利用巴斯德畢赤酵母表達綿羊卵泡促激素發現,因糖基化影響其表達量僅為0.1 μg/L。畢赤酵母表達系統表達出的丹酚酸酯酶量不高,推測這可能與外源基因整合到畢赤酵母上的拷貝數過低有關。Sreekrishna[24]等的研究顯示,整合了20個腫瘤壞死因子基因在染色體上的畢赤酵母,其重組子表達水平是單拷貝重組子的200倍。具體影響畢赤酵母中丹酚酸酯酶表達水平的因素還需今后進一步研究。
3 結論
本論文成功構建了應用于丹酚酸酯酶異源表達的兩種表達體系:大腸桿菌表達體系與畢赤酵母表達體系。利用大腸桿菌表達系統能夠誘導丹酚酸酯酶高效表達,并且外源基因在單獨表達時的表達量占菌體總蛋白量較高,部分可能形成包涵體,酶活力喪失;利用畢赤酵母表達體系可以使蛋白分泌性表達,所共表達的重組蛋白具有的一定的丹酚酸酯酶的催化活性,但表達量不理想。
由此可見,畢赤酵母雖然是真核表達系統,可以進行蛋白質的翻譯后加工及修飾,可能更利于表達真核生物的蛋白,但不同的外源蛋白在同一表達系統中的表達量卻存在著千差萬別。大腸桿菌表達系統能夠使丹酚酸酯酶高效表達,并且形成的包涵體富含重組蛋白,尋求到適合的體外復性條件后,大腸桿菌表達系統將會是大量生產重組蛋白的最有效途徑之一,隨著結構生物學、蛋白質工程學及相關新技術及設備的發展完善,設計最佳的該酶的包涵體變復性方案將成為可能。大腸桿菌表達體系還具有培養周期短、成本低、高效表達和易于純化等優點,也使其更適合于生產中應用[25]。因此,大腸桿菌體系更為適合丹酚酸酯酶的大量表達,對研究利用工程菌生物合丹酚酸酯酶具有指導意義。
[1]Chao C,Yin Y,Duan J,et al. Neuroprotective effect and underlying mechanism of sodium danshensu[3-(3,4-dihydroxyphenyl)lactic acid from Radix and Rhizoma Salviae miltiorrhizae=Danshen]against cerebral ischemia and reperfusion injury in rats[J]. Phytomedicine,2015,22(2):283-289.
[2]Zhao G R,Zhang H M,Ye T X,et al. Characterization of the radical scavenging and antioxidant activities of danshensu and salvianolic acid B[J]. Food and Chemical Toxicology,2008,46(1):73-81.
[3]陳瑞戰,張守勤,張永宏,等. 超高壓提取丹參素的研究[J]. 農業工程學報,2008,24(1):291-295.
[4]鄒曉軍,高小利. 丹參中丹參素和原兒茶醛的微波提取工藝研究[J]. 中藥材,2007,30(6):735-737.
[5]王金軍,林琳,李景令,等. 丹酚酸 B 水解生產丹參素的工藝優化[J]. 天然產物研究與開發,2015,27:715-719.
[6]關丹,高建梅,王何,等. 丹參中水溶性成分提取新方法的研究[J]. 大連輕工業學院學報,2007,26(3):193-195.
[7]于翀,王瑩,魚紅閃,等. 丹酚酸B酶產物的分離[J]. 大連工業大學學報,2009,28(3):161-164.
[8]馬明飛,李樹金,金鳳燮,等. 人參皂苷葡萄糖苷酶基因在大腸桿菌中的表達及包涵體的變復性[J]. 大連工業大學學報,2012,31(1):8-11.
[9]Laemmli U K. Cleavage of structural proteins during the assemble of the head of bacteriophage T4[J]. Nature,1970,227:680-685.
[10]劉俊梅,胡耀輝. 薄層層析在多元醇檢測中的應用[J]. 食品科技,2007,6:224-226.
[11]Simone,Vincenzi,Andrea,et al. Anomalous electrophoretic behavior of a chitinase isoform from grape berries and wine in glycol chitin-containing sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels[J]. Electrophoresis,2005,26(1):60-63.
[12]Richards J P,B?chinger H P,Goodman R H,et al. Analysis of the structural properties of cAMP-responsive element-binding protein(CREB)and phosphorylated CREB[J]. Journal of Biological Chemistry,1996,271(23):13716-13723.
[13]Lucía,Vivian,Antonio,et al. Anomalous electrophoretic behavior of a very acidic protein:ribonuclease U2[J]. Electrophoresis,2005,26:3407-3413.
[14]Armstrong D J,Roman A. The anomalous electrophoretic behavior of the human papillomavirus type 16 E7 protein is due to the high content of acidic amino acid residues[J]. Biochemical and biophysical research communications,1993,192(3):1380-1387.
[15]侯穎. rhddADAM15抗腫瘤及抗血管新生作用研究[D]. 無錫:江南大學,2014.
[16]Yang W,Zhang L,Lu Z,et al. A new method for protein coexpression in Escherichia coli using two incompatible plasmids[J]. Protein expression and purification,2001,22(3):472-478.
[17]Grosjean H,Fiers W. Preferential condon usage in prokaryotic genes:the optimal condon-anticondon interaction energy and the selective condon usage in efficiently expression genes[J]. Gene,1982,18(3):199-209.
[18]李單單. 糖苷水解酶GH12家族活性架構中關鍵氨基酸的功能分析[D]. 威海:山東大學,2013.
[19]Large T H,Rauh J J,De Mello,et al. Two molecular weight forms of muscarinic acetylcholine receptors in the avian central nervous system:switch in predominant form during differentiation of synapses[J]. Proceedings of the National Academy of Sciences of the United States of America,1985,82(24):8785-8789.
[20]Schlegl R,Iberer G,Machold C,et al. Continuous matrix-assisted refolding of protein[J]. Journal of Chromatography A,2003,1009(1-2):119-132.
[21]Vallejo L F,Rinas U. Strategies for the recovery of active proteins through refolding of bacterial inclusion body proteins[J]. Microbial Cell Factories,2004,3(1):85-100.
[22]Jungbauer A,Kaar W,Schlegl R. Folding and refolding of proteins in chromatographic beds[J]. Current Opinion in Biotechnology,2004,15(5):487-494.
[23]Fidler A E,Lin J S,Chie WN,et al. Production of biologically active tethered ovine FSH beta alpha by the methylotrophic yeast Pichia pastoris[J]. Mol Endocrinol,2003,30(2):213-225.
[24]Sreekrishna K,Nelles L,Potenz R,et al. High-level expression,purification,and characterization of recombinant human tumornecrosis factor synthesized in the methy lotrophic yeast Pichia pastoris[J]. Biochem istry,1989,28(9):4117-4125.
[25]Baneyx F. Recombinant protein expression in Escherichia coli[J]. Current opinion in biotechnology,1999,10(5):411-421.
Vector construction and biotransformation of double subunits salvianolic acid-esterase
CHENG Yan1,LIU Chun-ying1,YL Taehoo2,YU Hong-shan1,*
(1.School of Biological Engineering,Dalian Polytechnic University,Dalian 116034,China;2.Department of Oriental Medicinal Material and Processing,College of Life Science,Kyung Hee University,Yongin 446-701,Korea)
In order to achieve the salvianolic acid-esterase expressinvitro,the salvianolic acid-esterase were heterologous expressed byE.coliandPichiapasiorisrespectively. TheE.coilsystem constructed expression vector with pET 28a and pET 32a and carried out induced expression and coexpression of the recombinantE.coil. ThePichiapastorissystem constructed coexpression vector with pPIC9K and carried out induced expression of the recombinantPichiapastoris. The research found that the salvianolic acid-esterase were induced inE.coliandPichiapasioris. The protein was highly expressed inE.colisystem,but it did not exhibit the enzyme activity. The salvianolic acid-esterase were expressed inPichiapasioris,and the expression product had salvianolic acid-esterase activity. But the quantity of expression was not high. It showed that theE.coliexpression system was more suitable for the salvianolic acid-esterase and provided a exogenous expression method of salvianolic acid-esterase.
salvianolic acid-esterase;E.coil;Pichiapastoris;cloning and expression
2016-01-12
程妍(1991-),女,碩士研究生,研究方向:天然產物的生物轉化,E-mail:13478610863@163.com。
魚紅閃(1968-),男,博士,教授,研究方向:天然產物的生物轉化,E-mail:hongshan@dlpu.edu.cn。
“重大新藥創制”科技重大專項(2012ZX09503001-003);國家外專局高端外國專家項目(GDT20152100019)。
TS201.2
A
1002-0306(2016)13-0143-06
10.13386/j.issn1002-0306.2016.13.020
