不同類型白癜風基因譜表達差異的初步研究
2016-09-10 09:14:20聶慧瓊張小燕侯秀麗邵瓊琰
安徽醫科大學學報 2016年5期
聶慧瓊,王 平,,張小燕,侯秀麗,許 文,邵瓊琰
不同類型白癜風基因譜表達差異的初步研究
聶慧瓊1,王平1,2,張小燕2,侯秀麗1,許文2,邵瓊琰3
目的 探討白癜風臨床類型與基因表達差異的相關性。方法 分別選擇節段型白癜風(SV)、非節段型白癜風(NSV)和健康對照(HI)各4例,應用Phalanx人全基因組表達譜芯片檢測外周血淋巴細胞基因譜表達差異,并通過RTPCR進行驗證。結果 與HI相比,SV有239個上調基因和175個下調基因,主要涉及適應性免疫反應、細胞因子受體相互作用、趨化因子信號通路,NSV有88個上調基因和560個下調基因,涉及先天性免疫、細胞自噬與凋亡、黑素細胞生物學、泛素介導的蛋白裂解與酪氨酸酶代謝。SV和NSV共同基因表達差異包括60個上調基因和60個下調基因;相對于SV,NSV表達223個上調基因和129個下調基因,主要包括嘌呤、嘧啶、鞘脂類代謝。結論 SV有不同于NSV的遺傳背景與發病機制。
白癜風,節段型;白癜風,非節段型;基因表達
網絡出版時間:2016-4-19 11:04:48 網絡出版地址:http://www.cnki.net/kcms/detail/34.1065.R.20160419.1104.044.html
白癜風是一種常見的皮膚黏膜色素脫失性疾病,其病因和確切發病機制至今尚不完全清楚,涉及遺傳、免疫炎癥、神經體液、細胞毒性、氧化應激、功能性黑素細胞凋亡和(或)丟失及環境因素等假說[1-2]。根據皮損分布特點,白癜風分為兩種類型,節段型白癜風(segmental vitiligo,SV):白斑沿某一皮神經節段支配的皮膚區域走向分布,通常皮損不超過中線;非節段型白癜風(non-segmental vitiligo,NSV):又稱尋常型白癜風,為廣泛、局灶、或散在白斑。SV和NSV不僅在皮損分布上各具特點,對治療選擇與治療反應也存在較大差異,兩者存在不同的病理機制[3-4]。該研究采用高通量的基因芯片技術,篩選SV和NSV患者外周血差異表達基因,探討不同類型白癜風發病相關基因的差異,為進一步揭示白癜風發病機制和采用個體化治療方案提供新思路。
1 材料與方法
1.1標本收集 選取杭州市第三人民醫院皮膚科SV和NSV各4例(表1),病例均有典型白斑分布特點,并通過Wood燈和激光共聚焦顯微鏡再次確診。患者均無伴發其他器質性疾病、斑禿、橋本甲狀腺炎等自身免疫性疾病及感染性疾病。入選者均為初次診治,均未系統應用糖皮質激素、免疫抑制劑、光敏劑及紫外線治療。兩組的性別、年齡及病程差異均無統計學意義,有可比性。健康對照組均來自我院健康體檢者。本研究獲得杭州市第三人民醫院醫學倫理委員會批準,入選病例均簽署知情同意書。
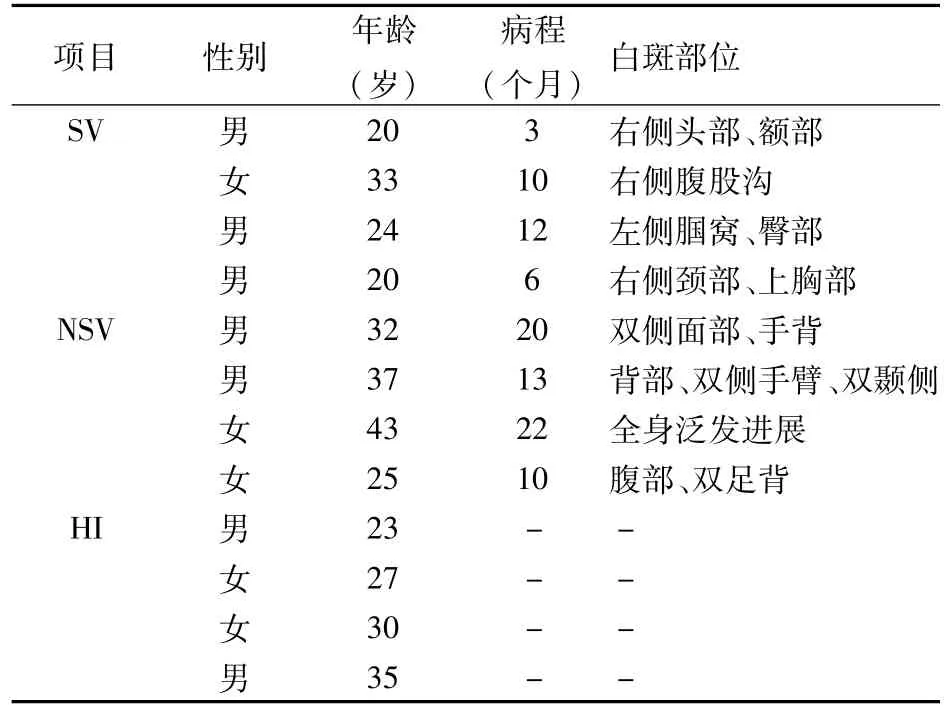
表1 病例資料
1.2全基因表達芯片檢測 抽取淺靜脈抗凝血6 ml,常規分離外周血淋巴細胞,TRIzol抽提RNA后-20℃保存,送上海儀方生物技術有限公司進行Phalanx人全基因組表達譜芯片(人32 679個基因)檢測。每組重復檢測4次。基因芯片數據利用Rosetta Resolver軟件(USA)進行處理,篩選全基因組表達譜芯片獲取的基因差異表達水平≥0.585,且經T-test統計分析顯著差異表達的基因(P<0.05)判定為差異表達基因。
1.3RT-PCR驗證 選取6條基因產物進行RTPCR驗證,其引物序列見表2。操作步序按常規操作,應用2-ΔΔCt法計算相對表達量。

表2 RT-PCR引物設計
1.3統計學處理 采用SPSS 13.0統計軟件進行分析,數據以 ˉx±s表示,組間比較采用兩樣本 t檢驗;系統聚類分析法將差異性表達基因數據進行對數變換,應用Version3.0軟件進行數據分析。
2 結果
2.1基因芯片檢測結果
2.1.1基因表達情況 相對于HI,SV中239個基因表達上調,175個表達下調,差異性表達基因發生聚類(圖1A)。利用基因本體論(gene ontology,GO)數據庫提供的3種本體論(生物過程、細胞組分、分子功能)對差異表達基因進行GO分類,生物學過程中(表3),主要涉及以下3個富集項目:信號轉導、生物合成過程及細胞營養代謝過程;細胞組分中(表4),主要是25%的細胞質,19%細胞核,12%細胞支架等;分子功能中(表5),差異性表達基因主要分為離子的結合、脫氧核糖核酸(DNA)的結合、信號轉導活性、酶的結合。這些差異性表達的基因主要作用的通路包括細胞因子-細胞因子受體相互作用,腫瘤通路、趨化因子信號通路、MAPK通路(表6)。
2.1.2NSV基因表達情況 相對于HI,NSV表達688個上調基因和560個下調基因。通過用聚類cluster軟件對差異表達的基因進行聚類分析的結果顯示分析(圖1B),左側探針聚類區主要分上調和下調基因,這些基因參與多種功能如免疫反應,信號通路,轉錄因子。通過GO分析,生物學過程中(表3),主要富集項目包括轉錄調節,信號轉導,固有免疫反應,氧化還原過程;細胞組分中(表4),24%細胞膜,20%細胞核,19%細胞液等物質參與此過程;分子功能中(表5),差異性基因包含脫氧核糖核酸(DNA)的結合,受體轉錄因子、轉移酶、GTP酶活性。通過KEGG通路分析,這些差異性表達基因主要涉及的通路如下:MAPK信號通路,Toll樣受體信號通路、Jak-STAT信號通路,趨化因子通路、泛素水解蛋白及C-C受體反應(表6)。
2.1.3SV和NSV基因表達差異比較 相對于HI,SV和NSV共同表達60個上調節基因和60個下調基因。相對于SV,NSV表達223個上調基因和129個下調基因。聚類分析差異性表達基因主要分為3種類型(圖1C)。通過 GO分析,生物學過程中(表3),主要分為信號轉導、生物合成過程細胞分化和免疫系統過程;細胞組分中(表4),蛋白復合物,質膜,細胞漿、細胞核和細胞液占75%以上;分子功能中(表5),包含19%信號轉導蛋白活性,16%酶的結合和10%氧化還原酶活性。基因通路分析顯示,這些差異性表達基因參與的過程包括嘌呤、嘧啶、鞘脂類和色氨酸代謝,免疫適應(表6)。
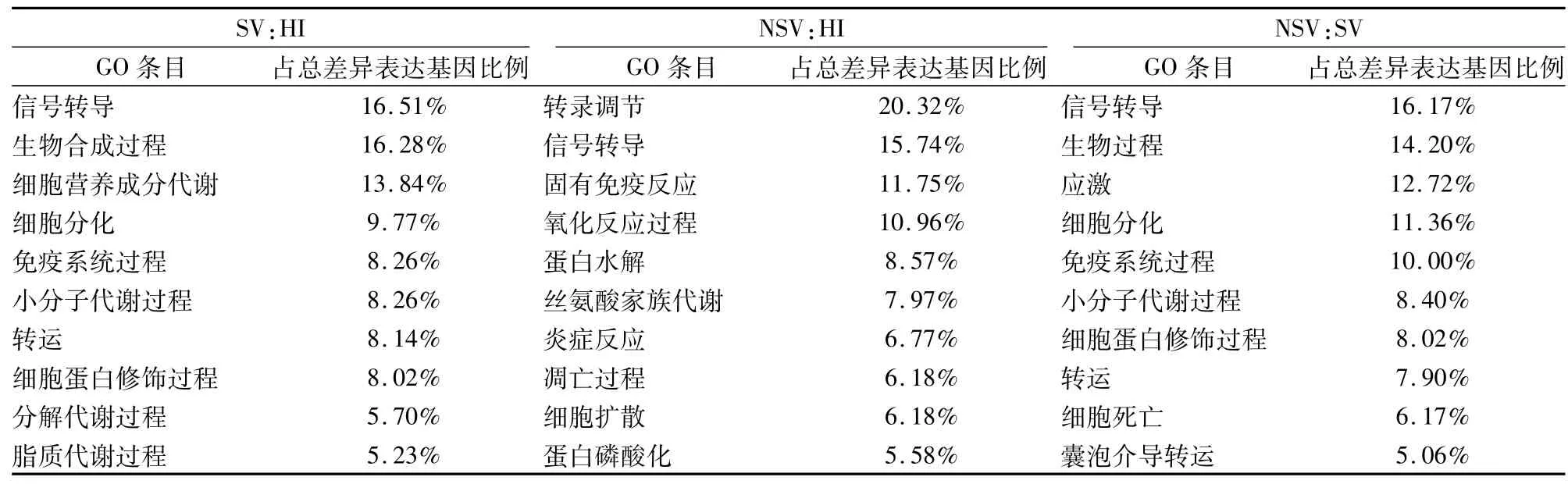
表3 主要生物學過程 GO分析結果
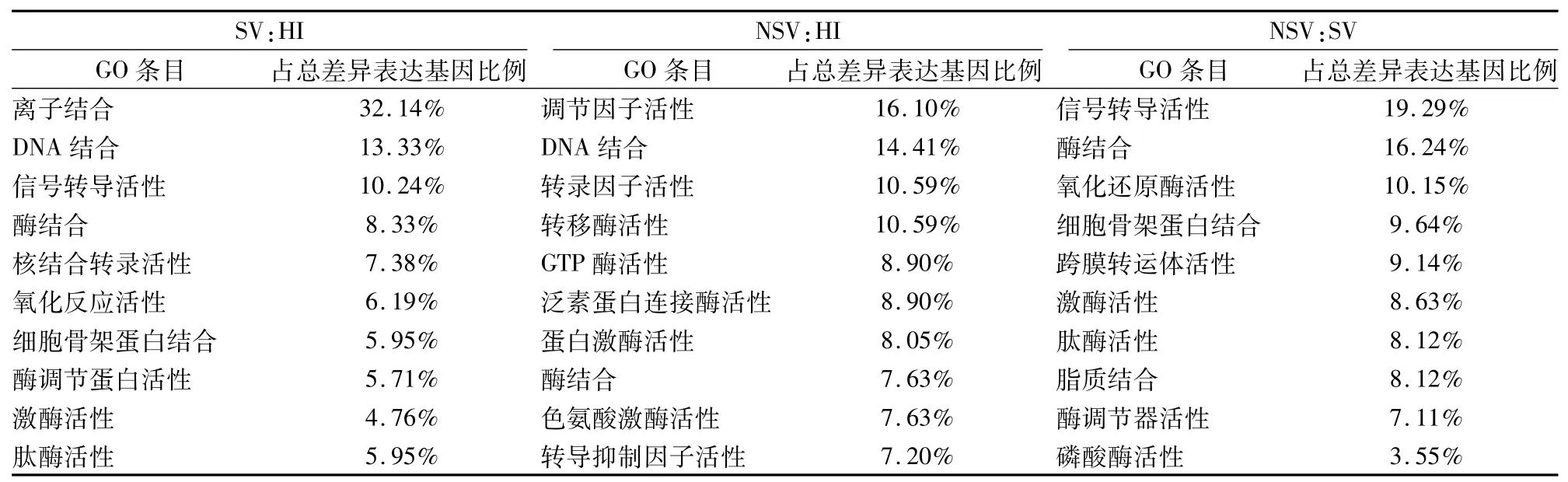
表5 主要分子功能 GO分析結果
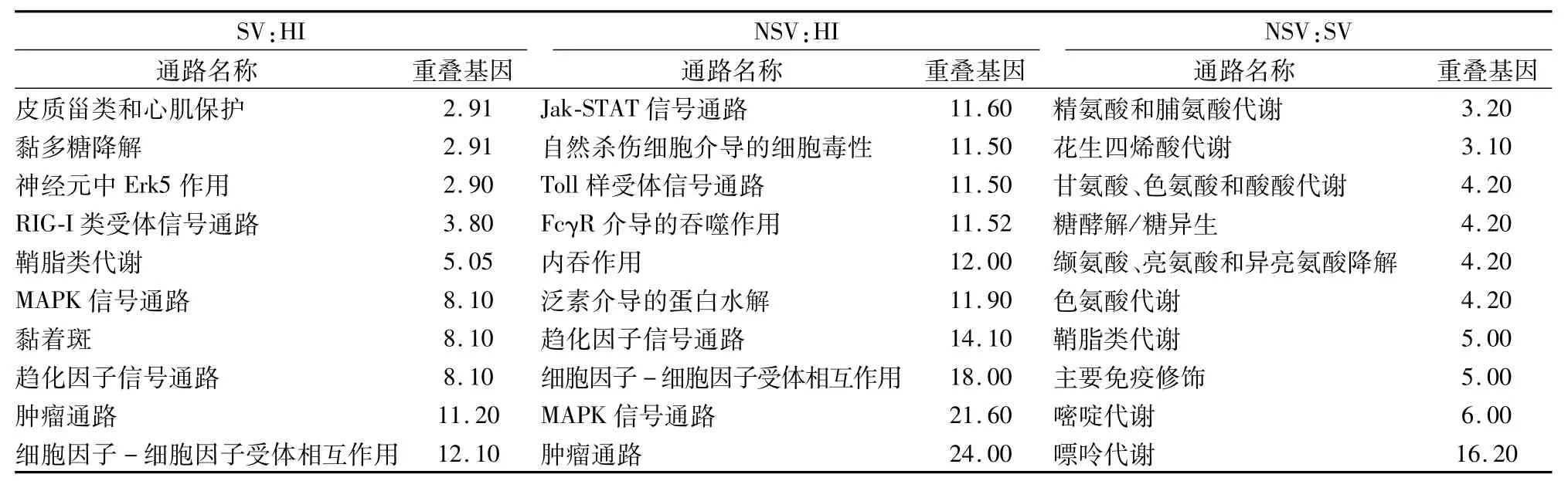
表6 差異表達基因主要pathway通路分析結果
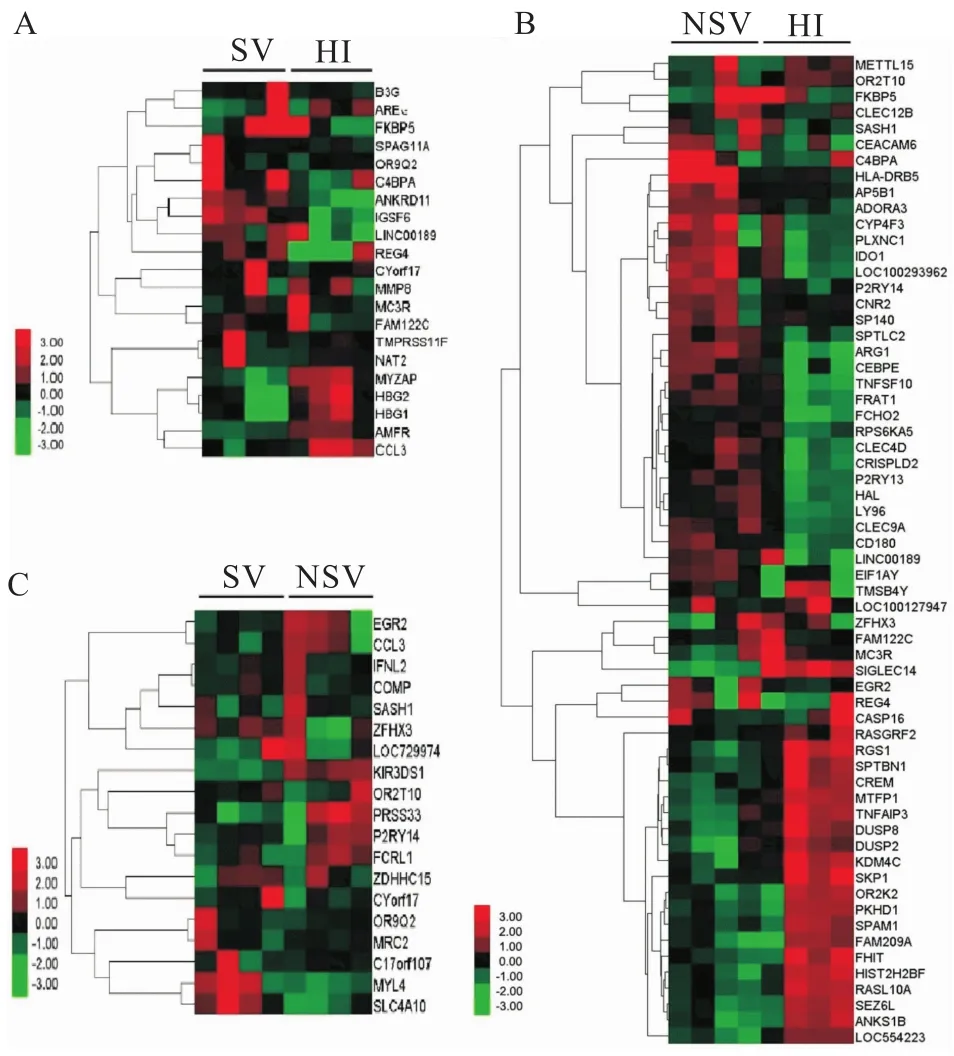
圖1 聚類分析A:SV和HI前5%差異性表達基因聚類分析;B:NSV和HI前5%差異性表達基因聚類分析;C:SV和NSV5%差異性表達基因聚類分析
2.2RT-PCR結果驗證 RT-PCR試驗驗證6條基因產物見表2。基因芯片與RT-PCR檢測結果一致(圖2)。相對于 HI,NSV中 EGR2、UBE2D1、KIR3DS1的mRNA表達上調;SV中,UBE2D1、MMP8表達上調,CCL3、KIR3DS1和MRC2表達下調;相對于SV,NSV中EGR2、CCL3和KIR3DS1的mRNA表達上調,MMP8表達下調。
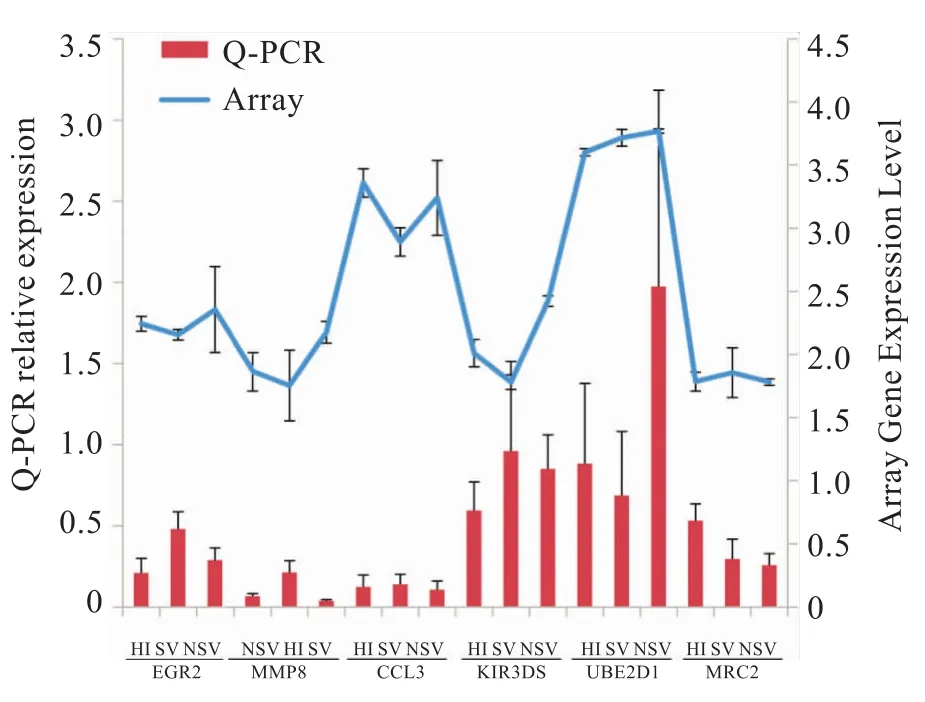
圖2 RT-PCR驗證對比基因芯片結果
3 討論
免疫反應在白癜風發病機制中起著重要作用,既往研究[5]表明,白癜風患者存在著體液免疫、細胞免疫、細胞因子及其受體功能的異常以及自身免疫耐受狀態的破壞。本研究結果表明,SV、NSV及HI均有涉及Toll樣受體信號通路,B細胞和T細胞分化,淋巴細胞增殖,免疫反應激活等免疫反應。但是,SV主要是適應性免疫反應起著重要作用,而NSV則是固有免疫反應及B細胞分化、活化占主導作用,提示兩種類型白癜風的黑素細胞破壞過程可能由不同免疫過程參與。
Jeong et al[6]對225名NSV和439名健康對照組進行單核苷酸多態性(SNP)研究,發現自噬相關基因紫外線輻射抵抗相關基因(ultraviolet radiation resistance-associated gene,UVRAG)多態性和NSV密切相關,推測 UVRAG可能和 NSV易感性相關。本研究則有類似結果,即NSV與自噬相關性較更高。相對于HI,NSV存在14個自噬相關的差異性表達基因,而只有一個自噬相關基因PAFAH1B2有差異性表達,提示自噬可能在NSV的病理機制中起著重要作用。
過量的氧化應激產物可通過對黑素細胞的直接毒性作用、干擾表皮細胞生物嘌呤代謝、抑制細胞黑素合成酶功能等多種途徑,損傷或破壞黑素細胞,影響黑素代謝,甚至導致黑素細胞的死亡[7]。在進展期白癜風患者中,外周血單核細胞超氧化歧化酶活性增強,谷胱甘肽和維他命E水平減低[8]。本研究顯示兩組白癜風均存在氧化酶活性相關基因如SOD1和SOD2表達上調,表明氧化應激在SV和NSV發病中作用相當。
功能性黑素細胞凋亡和(或)丟失,是白癜風發病機制的假說之一[9-10]。白癜風患者受損皮膚中,可發現凋亡角質形成細胞,且抗凋亡蛋白Bcl-2、FLIP低表達,而凋亡多肽抗原和P53則高表達[10]。本研究中,凋亡相關基因多高表達在NSV,而SV幾乎不表達,提示NSV與細胞凋亡關系密切。
黑素細胞產生黑素受酪氨酸酶相關家族調控,包括酪氨酸酶和酪氨酸酶相關蛋白,其受小眼畸形轉錄因子調控[11]。本研究表明,NSV有45個酪氨酸代謝和酪氨酸激酶啟動子活性相關基因差異表達,而不見于SV,推測NSV存在酪氨酸代謝異常。另外,NSV還存在幾種參與泛素介導的蛋白水解差異性表達基因。這些基因可能參與NSV中相關蛋白異常水解過程,從而影響其黑素細胞功能。
綜上所述,本研究顯示SV和NSV均存在免疫異常和高氧化應激反應過程,而相對于SV,NSV特異性表達自噬、凋亡、蛋白水解和酪氨酸酶代謝相關基因,提示兩者有不同的遺傳背景和發病機制。本研究對可能與白癜風相關的6個基因進行了RTPCR驗證,驗證結果顯示與基因芯片結果相符,這也從另一方面說明了基因芯片結果的可信度。期待今后多環境多樣本交叉研究資料,為SV和NSV靶基因篩選及靶向治療提供分子基礎。
[1] Passeron T,Ortonne JP.Physiopathology and genetics of vitiligo[J].JAutoimmun,2005,25:63-8.
[2] Guerra L,Dellambra E,Brescia S,et al.Vitiligo:pathogenetic hypotheses and targets for current therapies[J].Curr Drug Metab,2010,11(5):451-67.
[3] 王 平,洪為松,章莉,等.白癜風表皮黑素細胞超微結構及小眼畸形相關轉錄因子轉錄調控研究[J].中華皮膚科雜志,2010,43(7):37-40.
[4] 倪亞杰,王平,洪為松,等.小眼畸形相關轉錄因子轉錄調控與白癜風臨床類型的相關性研究[J].醫學研究雜志,2013,42(3):52-5.
[5] Le Poole IC,Luiten R M.Autoimmune etiology of generalized vitiligo[J].Curr Dir Autoimmun,2008,10:227-43.
[6] Jeong T J,Shin M K,Uhm Y K,et al.Association of UVRAG polymorphismswith susceptibility to non-segmental vitiligo in a Korean sample[J].Exp Dermatol,2010,19(8):e323-5.
[7] Glassman S J.Vitiligo,reactive oxygen species and T-cells[J]. Clin Sci(Lond),2011,120(3):99-120.
[8] Dell'Anna M L,Maresca V,Briganti S,etal.Mitochondrial impairment in peripheral blood mononuclear cells during the active phase of vitiligo[J].J Invest Dermatol,2001,117(4):908-13.
[9] Moretti S,Fabbri P,Baroni G,et,al.Keratinocyte dysfunction in vitiligo epidermis:cytokinemicroenvironment and correlation tokeratinocyte apoptosis[J].HistolHistopathol,2009,24(7):849-57.
[10]Lee A Y,Youm Y H,Kim N H.Keratinocytes in the depigmented epidermis of vitiligo aremore vulnerable to trauma(suction)than keratinocytes in the normally pigmented epidermis,resulting in their apoptosis[J].Br JDermatol,2004,151(5):995-1003.
[11]Murisier F,Beermann F.Genetics of pigment cells:lessons from the tyrosinase gene family[J].Histol Histopathol,2006,21(5):567-78.
Prelim inary study of differential gene expression profiling between segmental vitiligo and non-segmental vitiligo
Nie Huiqiong1,Wang Ping1,2,Zhang Xiaoyan2,et al
(1Clinical College of Hangzhou,Anhui Medical University,Hangzhou 310000;2Dermatological Dept of Hangzhou Third People's Hospital,Hangzhou 310000)
Objective To investigate the correlation between clinical pattern and differential gene expression in patientswith vitiligo.Methods Peripheral blood lymphocytes obtained from four cases of segmental vitiligo(SV),non-segmental vitiligo(NSV)and healthy individual(HI)respectively.Whole genome expression microarrays were used to assay the gene expression profiles between SV,NSV and HI.Quantitative PCR assay was used to validate the gene expression of array.Results Compared to HI,239 over-expressed and 175 down-expressed genes were detected in SV,which weremainly involved in the adaptive immune response,cytokine-cytokine receptor interaction and chemokine signaling.In NSV,88 over-expressed and 560 down-expressed geneswere found and were mainly involved in the innate immune,autophagy,apoptosis,melanocyte biology,ubiquitin mediated proteolysis and tyrosine metabolism,which were different from SV.Sixty over-expressed and sixty down-expressed genes shared similar tendency in SV and NSV.Compared to SV,223 over-regulated and 129 down-regulated geneswere found in NSV,which weremainly involved in themetabolism of purine,pyrimidine and sphingolipid.Conclusion Theremay exit different genetic background and pathogenesis between SV and NSV.
segmental vitiligo;non-segmental vitiligo;gene expression
R 75
A
1000-1492(2016)05-0707-05
2016-02-22接收
杭州市醫學重點專科專病項目(編號:20140733Q19);浙江省醫藥衛生科技計劃項目(編號:2014KYB200)
1安徽醫科大學附屬杭州臨床學院皮膚科,杭州 310000
2杭州市第三人民醫院皮膚科,杭州 310000
3浙江中醫藥大學附屬杭州市第三人民醫院皮膚科,杭州
310000
聶慧瓊,女,碩士研究生;
王 平,男,主任醫師,碩士生導師,責任作者,E-mail:dermwang@aliyun.com
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中國特種設備安全(2018年11期)2019-01-08 02:08:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
