LCMSMS法測(cè)動(dòng)物源食品中硝呋烯腙殘留量的不確定度評(píng)定
2016-09-02 03:22:11羅艷霞
廣州化工 2016年13期
關(guān)鍵詞:標(biāo)準(zhǔn)
羅艷霞
(上海天祥質(zhì)量技術(shù)服務(wù)有限公司,上海 200233)
?
LCMSMS法測(cè)動(dòng)物源食品中硝呋烯腙殘留量的不確定度評(píng)定
羅艷霞
(上海天祥質(zhì)量技術(shù)服務(wù)有限公司,上海200233)
建立了液相色譜-串聯(lián)質(zhì)譜測(cè)定動(dòng)物源食品硝呋烯腙的方法,并對(duì)其不確定度進(jìn)行了評(píng)定。本文研究了液相色譜-串聯(lián)質(zhì)譜測(cè)定動(dòng)物源食品硝呋烯腙含量的方法,并依據(jù)CNAS-GL06-2006《化學(xué)分析中不確定度的評(píng)估指南》和JJF1059.1-2012《測(cè)量不確定度評(píng)定與表示》規(guī)定的原理與方法,分析了實(shí)驗(yàn)過(guò)程中不確定性的來(lái)源,建立了不確定度評(píng)定的數(shù)學(xué)模型,計(jì)算并分析了合成不確定度的結(jié)果。由結(jié)果可知:在液相色譜-串聯(lián)質(zhì)譜測(cè)定動(dòng)物源食品硝呋烯腙含量過(guò)程中,校準(zhǔn)曲線擬合與標(biāo)準(zhǔn)溶液的配置是影響其不確定度的主要因素,液相色譜-串聯(lián)質(zhì)譜儀和測(cè)量重復(fù)性是次要因素。
LCMSMS;動(dòng)物源性食品;硝呋烯腙;不確定度
我國(guó)是畜牧業(yè)大國(guó),隨著人們對(duì)動(dòng)物源食品由需求型向質(zhì)量型的轉(zhuǎn)變,動(dòng)物源食品中的獸藥殘留已逐漸成為全世界關(guān)注的一個(gè)焦點(diǎn)[1]。目前在養(yǎng)殖過(guò)程中,普遍濫用藥物添加劑或高效抗生素等成為影響動(dòng)物源性食品安全的主要原因。隨著人們對(duì)食品與環(huán)境質(zhì)量要求的不斷提高及對(duì)抗生素認(rèn)識(shí)的不斷深入,飼料中抗生素的添加所引起的問(wèn)題愈來(lái)愈引起人們的關(guān)注。
硝呋烯腙 (Nitrovin)是20世紀(jì)70年代中期發(fā)現(xiàn)的一種新型抗生素,它是一種呋喃類化學(xué)合成藥物,它通過(guò)增加動(dòng)物腸粘膜的通透性,從而能有效地提高營(yíng)養(yǎng)物質(zhì)在動(dòng)物消化道中的消化和吸收;還能夠有效抑制動(dòng)物消化道內(nèi)病原微生物對(duì)葡萄糖的利用,使其新陳代謝過(guò)程受阻,從而抑制和殺滅多種病原微生物,減少動(dòng)物感染疾病的機(jī)會(huì),提高抗病能力[2]。由于抗生素的耐藥性、殘留和二重感染的特性,所帶來(lái)的弊端和危害已成為人類所面臨的重大挑戰(zhàn)。中華人民共和國(guó)農(nóng)業(yè)部國(guó)家藥品監(jiān)督管理局公告(第193號(hào)):食品動(dòng)物禁用的獸藥及其它化合物清單明確指出硝呋烯腙禁止使用。因此,研究建立動(dòng)物源性食品中硝呋烯腙的殘留檢測(cè)方法具有重要意義。
目前有關(guān)檢測(cè)動(dòng)物源性食品中的硝呋烯腙殘留量的方法有高效液相色譜法[3],高效液相質(zhì)譜聯(lián)用法[4-5]等少量的文獻(xiàn)報(bào)道,國(guó)家也尚未制定相關(guān)的檢測(cè)標(biāo)準(zhǔn)。本文建立了硝呋烯腙的高效液相色譜-串聯(lián)質(zhì)譜檢測(cè)方法,其中硝呋烯腙色譜峰保留時(shí)間是3.47min, 線性范圍是1.0~50ng/mL,相關(guān)系數(shù)R為0.999。該方法實(shí)驗(yàn)過(guò)程簡(jiǎn)單,有利于食品檢測(cè)機(jī)構(gòu)的批量檢驗(yàn)。并且依據(jù)CNAS-GL06-2006《化學(xué)分析中不確定度的評(píng)估指南》[6]和JJF1059.1-2012《測(cè)量不確定度評(píng)定與表示》[7]規(guī)定的原理與方法,對(duì)本文測(cè)試方法的準(zhǔn)確可靠性進(jìn)行了詳細(xì)的分析,用合成不確定度[8-13]為測(cè)量結(jié)果提供理論基礎(chǔ)和科學(xué)依據(jù)。
1 實(shí) 驗(yàn)
1.1材料
1.1.1試劑
硝呋烯腙標(biāo)準(zhǔn)品,Dr.Ehrenstorfer; 乙腈(色譜純),F(xiàn)isher;甲醇(色譜純),F(xiàn)isher;甲酸(分析純),國(guó)藥;二甲基甲酰胺(分析純),國(guó)藥。
1.1.2儀器
AB4500型高效液相色譜-串聯(lián)質(zhì)譜檢測(cè)儀,ABSCIEX公司;FE20K型酸度計(jì)、MS204S型電子天平、XS105型電子天平,瑞士梅特勒托利多;SF-DL-4M型高速冷凍離心機(jī),上海菲恰爾分析儀器公司;SK250H型超聲振蕩儀,上海科導(dǎo)超聲儀器有限公司。
1.2實(shí)驗(yàn)方法
1.2.1標(biāo)準(zhǔn)溶液的配制
精密稱取10.00mg硝呋烯腙標(biāo)準(zhǔn)品,置于100mL容量瓶中,加入2mL二甲基甲酰胺溶解,再用甲醇定容,混勻,得到標(biāo)準(zhǔn)儲(chǔ)備溶液。用單標(biāo)線刻度為1mL的移液管準(zhǔn)確移取0.1mL標(biāo)準(zhǔn)儲(chǔ)備溶液至100mL容量瓶中,甲醇釋至刻度,混勻,此標(biāo)準(zhǔn)中間液濃度為 100ng/mL;再用10mL刻度移液管分別移取0.1mL、0.5mL、1.0mL、2.0mL、5.0mL100μg/mL的標(biāo)準(zhǔn)中間液于5只10mL容量瓶中,甲醇釋至刻度,混勻,配置成的標(biāo)準(zhǔn)工作液濃度分別為1.0ng/mL、5.0ng/mL、10ng/mL、20ng/mL、50ng/mL(注:此處移液管、容量瓶均為A級(jí))。
1.2.2樣品預(yù)處理
精密稱取5.0000g樣品于50mL離心管中,用A級(jí)10mL刻度移液管吸取6mL二甲基甲酰胺加入離心管,振搖分散樣品,再加入9mL乙腈,超聲提取20min,4000r/min冷凍離心10min,過(guò)0.45和0.22μm有機(jī)膜,供LC-MS-MS分析測(cè)定。
1.2.3儀器條件
1.2.3.1液相色譜條件

表1 流動(dòng)相梯度條件Table 1 Mobile phase gradient conditions
色譜條件:色譜柱為AgilentC18(4.6x250mm, 5μm);進(jìn)樣量10μL;流速為0.3mL/min;柱溫為 30 ℃;流動(dòng)相A相甲醇,B相0.1%甲酸水溶液(見(jiàn)表1)。
1.2.3.2質(zhì)譜條件
離子源:電噴霧(ESI+)離子源;掃描方式:正離子掃描;檢測(cè)方式:多反應(yīng)監(jiān)測(cè)(MRM);電壓:5500V;霧化氣壓力:50psi;氣簾氣壓力:35psi;輔助氣壓力:50psi;碰撞氣壓力:9psi;離子源溫度:500 ℃;碰撞室入口電壓:10V;碰撞室出口電壓:6V;硝呋烯腙最佳碰撞電壓、碰撞能量等如表2所示。

表2 硝呋烯腙的離子對(duì)Table 2 The ion pair of nitrovin
1.3數(shù)學(xué)模型的建立[8-9]
樣品中硝呋烯腙含量計(jì)算公式如下:
式中: X——樣品中硝呋烯腙含量,μg/kg
C——樣品溶液中硝呋烯腙濃度,ng/mL
V——樣品定容體積,mL
m——樣品質(zhì)量,g
上述計(jì)算公式是由測(cè)量原理得到的,沒(méi)有考慮各種隨機(jī)因素對(duì)不確定度的影響,在此引入反映隨機(jī)影響的重復(fù)性系數(shù) frep,其數(shù)值等于1。評(píng)定不確定度的數(shù)學(xué)模型應(yīng)寫成如下形式:
1.4測(cè)量不確定度因素[8-13]
依據(jù)檢測(cè)過(guò)程和數(shù)學(xué)模型,不確定度的主要因素有:實(shí)驗(yàn)重復(fù)性條件產(chǎn)生的相對(duì)標(biāo)準(zhǔn)不確定度uref(frep);樣品稱樣量產(chǎn)生的相對(duì)標(biāo)準(zhǔn)不確定度uref(m);樣品定容體積引起的相對(duì)標(biāo)準(zhǔn)不確定度uref(V);標(biāo)準(zhǔn)溶液配制引起的相對(duì)標(biāo)準(zhǔn)不確定度uref(C);校準(zhǔn)曲線擬合引起的相對(duì)標(biāo)準(zhǔn)不確定度uref(Co); 液相色譜-串聯(lián)質(zhì)譜儀定量重復(fù)性產(chǎn)生的相對(duì)標(biāo)準(zhǔn)不確定uref(LCMSMS)。合成相對(duì)標(biāo)準(zhǔn)不確定度uref(X)計(jì)箅公式如下:
2 結(jié)果與討論
2.1實(shí)驗(yàn)重復(fù)條件下產(chǎn)生的相對(duì)標(biāo)準(zhǔn)不確定度uref(frep)
對(duì)樣品進(jìn)行7次重復(fù)測(cè)定所得到數(shù)據(jù)如表3所示。

表3 樣品測(cè)定結(jié)果Table 3 Samples determination results
重復(fù)測(cè)量7次的實(shí)驗(yàn)標(biāo)準(zhǔn)偏差為:
日常測(cè)量時(shí)為平行測(cè)定,結(jié)果是平均值,故平均值相對(duì)標(biāo)準(zhǔn)不確定度為:
實(shí)驗(yàn)重復(fù)條件下產(chǎn)生的標(biāo)準(zhǔn)偏差,考慮了所有的隨機(jī)影響。因此在分析之后的各因素時(shí),無(wú)需考慮隨機(jī)影響。
2.2樣品稱樣量的相對(duì)標(biāo)準(zhǔn)不確定度uref(m)
用天平稱取5.0000g樣品時(shí),因天平所允許的最大誤差為±0.0001g, 按矩形分布分析,稱樣時(shí)天平引入的相對(duì)標(biāo)準(zhǔn)不確定度為:
2.3樣品定容體積的標(biāo)準(zhǔn)不確定度uref(V)

2.4硝呋烯腙標(biāo)準(zhǔn)溶液的相對(duì)標(biāo)準(zhǔn)不確度uref(C)
2.4.1配制標(biāo)準(zhǔn)儲(chǔ)備液所引入的相對(duì)標(biāo)準(zhǔn)不確定度uref(C1)
(1)由硝呋烯腙標(biāo)準(zhǔn)品證書可知其純度為98.5%,由標(biāo)準(zhǔn)品純度引入的不確定度為:
(2) 準(zhǔn)確稱取標(biāo)準(zhǔn)品硝呋烯腙時(shí),天平所允許的最大誤差為±0.01mg, 按矩形分布分析,天平引入的相對(duì)標(biāo)準(zhǔn)不確定度為:

(4)因此配制標(biāo)準(zhǔn)儲(chǔ)備液時(shí)所引入的相對(duì)標(biāo)準(zhǔn)不確定度uref(C1)為:
2.4.2配制標(biāo)準(zhǔn)中間液所引入的相對(duì)標(biāo)準(zhǔn)不確定度uref(C2)
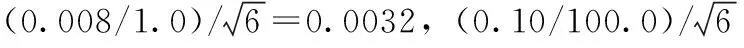
2.4.3配制標(biāo)準(zhǔn)工作液所引入的相對(duì)標(biāo)準(zhǔn)不確定度uref(C3)
依據(jù)JJG196-2006[14]常用玻璃量器檢定規(guī)程,據(jù)三角形分布分析,配制標(biāo)準(zhǔn)工作液所引入的相對(duì)標(biāo)準(zhǔn)不確定度uref(C3)見(jiàn)表4[9]。

表4 標(biāo)準(zhǔn)工作液配制引入的相對(duì)標(biāo)準(zhǔn)不確定度Table 4 The introduction of relative standard uncertainty of standard working liquid
①按三角分布分析,研討溶液溫度與校正時(shí)溫度差引起的體積不確定度0.00048,可知uref(VAi)為:
②按三角分布分析,研討溶液溫度與校正時(shí)溫度差引起的體積不確定度0.00048,可知uref(VBi)為:
③綜合以上得到相對(duì)標(biāo)準(zhǔn)不確定度uref(C3):
2.4.4配制硝呋烯腙標(biāo)準(zhǔn)溶液的相對(duì)標(biāo)準(zhǔn)不確定度uref(C)
2.5校準(zhǔn)曲線擬合引入的相對(duì)標(biāo)準(zhǔn)不確定度uref(Co)
用LCMSMS測(cè)定硝呋烯腙標(biāo)準(zhǔn)溶液5個(gè)濃度水平,可知相應(yīng)的峰面積,運(yùn)用最小二乘法擬合,擬合方程Y=aC+b和相關(guān)系數(shù)r(見(jiàn)表5)。

表5 測(cè)定及由擬合方程計(jì)算結(jié)果Table 5 Measurement and calculation results by fitting equations
注:a=35242,b=2406,r=0.999。
依據(jù)擬合的標(biāo)準(zhǔn)曲線,將樣品測(cè)定10次,得到的樣品溶液濃度測(cè)定結(jié)果表6所示。

表6 樣品溶液濃度測(cè)定結(jié)果Table 6 Sample solution concentration determination results
計(jì)算可知:C0=7.826ng/mL,X=23.48μg/kg
由標(biāo)準(zhǔn)曲線求濃度時(shí)的相對(duì)標(biāo)準(zhǔn)不確定度uref(C0)由下式計(jì)算[9]:
式中:a——標(biāo)準(zhǔn)工作曲線的斜率
P——重復(fù)測(cè)定樣品的數(shù)量
m——重復(fù)測(cè)定標(biāo)準(zhǔn)溶液的數(shù)量
n——配制的標(biāo)準(zhǔn)溶液數(shù)量
Sy/c——工作曲線的標(biāo)準(zhǔn)偏差
Sc/c——工作曲線各濃度點(diǎn)的離均差平方和
Yij——工作曲線各濃度點(diǎn)的測(cè)定響應(yīng)值
Yi——由線性方程計(jì)算所得響應(yīng)值
Ci——工作曲線各濃度點(diǎn)

C0——測(cè)試樣平均濃度
由以上公式計(jì)算的結(jié)果見(jiàn)表7。

表7 最小二乘法擬合標(biāo)準(zhǔn)曲線時(shí)引入的不確定度uref(C0)Table 7 Least squares fitting in uncertainty of standard curve
2.6液相色譜-串聯(lián)質(zhì)譜在測(cè)量過(guò)程中所引入的相對(duì)標(biāo)準(zhǔn)不確定度uref(LCMSMS)
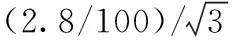
2.7合成相對(duì)標(biāo)準(zhǔn)不確定度及評(píng)定擴(kuò)展不確定度
2.7.1合成相對(duì)標(biāo)準(zhǔn)不確定度
依據(jù)文中各項(xiàng)不確定度分量(見(jiàn)表8),合成得到相對(duì)標(biāo)準(zhǔn)不確定度uref(X):
=0.0548

表8 各相對(duì)標(biāo)準(zhǔn)不確定度分量匯總表Table 8 Each relative standard uncertainty componentssummary table
2.7.2擴(kuò)展不確定度
當(dāng)包含因子k=2,置信水平約為95%,依據(jù)公式計(jì)算可得相對(duì)擴(kuò)展不確定度uref(X):
Uref(X)=uref(X)×k=0.1096
依據(jù)表6計(jì)算得,動(dòng)物源性樣品中硝呋烯腙測(cè)定的殘留量的最佳預(yù)估值為23.48μg/kg,故其擴(kuò)展不確定度U為:
U=23.48×Uref(X)=2.6μg/kg
3 結(jié) 論
本實(shí)驗(yàn)采用液相色譜-串聯(lián)質(zhì)譜法測(cè)定動(dòng)物源性樣品中硝呋烯腙殘留量,當(dāng)k=2,置信水平是95%時(shí),測(cè)得的硝呋烯腙含量為(23.48±2.6)μg/kg。本文中實(shí)驗(yàn)過(guò)程簡(jiǎn)單易行,用來(lái)對(duì)動(dòng)物源性食品中硝呋烯腙的測(cè)定準(zhǔn)確可靠性較高。
對(duì)實(shí)驗(yàn)過(guò)程及不確定度分量結(jié)果綜合分析可知:在液相色譜-串聯(lián)質(zhì)譜測(cè)定動(dòng)物源食品硝呋烯腙殘留量過(guò)程中,校準(zhǔn)曲線擬合與標(biāo)準(zhǔn)溶液的配置是影響其不確定度的主要因素,液相色譜-串聯(lián)質(zhì)譜儀和測(cè)量重復(fù)性是次要因素。
[1]郭曉娟.獸藥殘留產(chǎn)生的原因及危害[J].養(yǎng)殖技術(shù)顧問(wèn),2011,5(03):12-14.
[2]陳永豐,印遇龍,譚支良,等.硝呋烯腙對(duì)斷奶仔豬生產(chǎn)性能的影響[J].中國(guó)畜牧雜志,2003,39(03):29-30.
[3]LinSY,JengSL.High-performanceliquidchromatographicdeterminationofcarbadox,olaquindox,furazolidone,nitrofurazone,andnitrovininfeed[J].JournalofFoodProtection,2001,64(8):1231-1234.
[4]王金榮,李德發(fā),張麗英,等.硝呋烯腙的純化及其在飼料中的HPLC及LC-MS檢測(cè)技術(shù)研究[J].分析測(cè)試學(xué)報(bào),2006,26(z1):99-100.
[5]徐彥輝,何俊,顧亮,等. 液相色譜串聯(lián)質(zhì)譜法測(cè)定動(dòng)物源性食品中硝呋烯腙的殘留量[J].化學(xué)分析計(jì)量,2011,20(4):29-32.
[6]中國(guó)合格評(píng)定國(guó)家認(rèn)可委員會(huì),CNAS-GL06:2006化學(xué)分析中不確定度的評(píng)估指南[S].北京:中國(guó)計(jì)量出版社,2006.
[7]國(guó)家質(zhì)量技術(shù)監(jiān)督局.JJF1059.1-2012測(cè)量不確定度評(píng)定與表示[S].北京:中國(guó)計(jì)量出版社,2012.
[8]湯海靑,顧曉俊,殷居易,等.高效液相色譜法測(cè)定葡萄酒中苯甲酸含量的不確定度評(píng)定[J].食品工業(yè)科技,2013,34(15):295-298.
[9]煙利亞.高效液相色譜法測(cè)定淀粉中馬來(lái)酸含量的不確定度評(píng)定[J].廣州化工, 2014,42(20):145-148.
[10]蔡秋,朱明.高效液相色譜法測(cè)定茶飲料中苯甲酸結(jié)果不確定度評(píng)定[J].食品科學(xué),2007(02):277-280.
[11]汪輝,曹小彥,彭新凱,等.高效液相色譜法測(cè)定小麥粉與大米粉中甲醛次硫酸氫鈉含量的不確定度評(píng)定[J].食品科學(xué),2009,30(12):205-208.
[12]楊洋,徐春祥,車文軍.高效液相色譜法測(cè)定奶粉中的三聚氰胺及其不確定度分析[J].食品科學(xué),2010,31(04):250-253.
[13]朱佳,孫杰.液相色譜法測(cè)定果汁中糖精鈉的不確定度研究[J].食品研究與開(kāi)發(fā), 2012,33(02):156-158.
[14]國(guó)家質(zhì)量監(jiān)督檢驗(yàn)檢疫總局,JJG196-2006常用玻璃量器檢定規(guī)程[S].北京:中過(guò)國(guó)計(jì)量出版社,2006.
Uncertainty Evaluation for the Determination of Nitrovin Residues inAnimal-originFoodbyLiquidChromatographyTandemMassSpectrometry
LUO Yan-xia
(Shanghai Intertek Testing Services Co., Ltd., Shanghai 200233, China)
AliquidchromatographytandemmassspectrometrydeterminationofNitrovinresiduesinanimal-originfoodwasestablished,andthemeasurementuncertaintyofthismethodwasevaluated.Theliquidchromatography-tandemmassspectrometrydeterminationofanimaloriginfoodmethodofnitrovincontentwasstudied,accordingtotheprincipleandmethodinCNAS-GL06-2006GuidanceonEvaluatingtheUncertaintyinChemicalAnalysisandJJF1059.1-2012EvaluationandExpressionofUncertaintyinMeasurement,thesourceofuncertaintyintheexperimentprocesswasanalyzed,themathematicalmodelofuncertaintyevaluationwasestablished,theresultofthesynthesisofuncertaintywascalculatedandanalyzed.Theresultsshowedthatinthedeterminationofanimaloriginfoodofnitrovincontentintheprocess,calibrationcurvefittingandtheconfigurationofthestandardsolutionwerethemainfactorsaffectingtheuncertainty,liquidchromatography-tandemmassspectrometerandrepeatabilityweresecondaryfactors.
LCMSMS;animal-originfood;nitrovin;uncertainty
羅艷霞,女,本科,主要從事食品檢測(cè)工作。
O657.7+2
A
1001-9677(2016)013-0136-05
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當(dāng)代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動(dòng)漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(shù)(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質(zhì)量與標(biāo)準(zhǔn)化(2015年9期)2015-12-31 11:41:40
中國(guó)質(zhì)量與標(biāo)準(zhǔn)導(dǎo)報(bào)(2014年4期)2014-03-11 19:54:25
中國(guó)質(zhì)量與標(biāo)準(zhǔn)導(dǎo)報(bào)(2014年10期)2014-02-28 22:25:47
中國(guó)質(zhì)量與標(biāo)準(zhǔn)導(dǎo)報(bào)(2014年7期)2014-02-28 22:24:39
