溫室氣體氫氟烴的處理與利用
2016-06-22 13:46:47賈文志劉聰劉行石夢琦陳焱峰朱志榮
化工生產與技術 2016年4期
賈文志,劉聰,劉行,石夢琦,陳焱峰,朱志榮*
(1.湖北理工學院化學與化工學院,湖北黃石435003;2.同濟大學化學科學與工程學院,上海200092;3.上海三愛富新材料股份有限公司,上海200092)
氟化工
溫室氣體氫氟烴的處理與利用
賈文志1,劉聰1,劉行2,石夢琦1,陳焱峰3,朱志榮2*
(1.湖北理工學院化學與化工學院,湖北黃石435003;2.同濟大學化學科學與工程學院,上海200092;3.上海三愛富新材料股份有限公司,上海200092)
總結分析了近年來文獻和專利,將HFCs的處理與轉化方法分析歸納為消除處理與資源化轉化利用2大類,其中消除處理方法有氧化燃燒法、等離子法、催化水解法,資源化轉化利用方法有高溫裂解轉化烯烴法、共裂解法、催化脫氟化氫法。介紹了2大類、6種處理與轉化方法,并分析了不同的HFCs處理與轉化方法的利弊與工業應用前景。認為HFCs類物質可以轉化為附加值高、環境友好的含氟烯烴、含氟聚合物和碘氟烴等,是未來HFCs的資源化轉化利用的意義所在;而催化脫HF反應路線的技術關鍵問題是高效催化劑的開發。
氫氟烴;含氟烯烴;處理與轉化;催化脫氟化氫
20世紀70年代,美國科學家Molina首次提出氯氟烴(CFCs)類物質是造成大氣臭氧層空洞的主要物質(稱為ODS物質)[1]。CFCs物質破壞大氣臭氧層,給人類健康帶來嚴重的威脅。隨著科學技術的進步以及人們對環境保護日益重視,氟代烴類產品經歷了從CFCs到氫氟氯烴(HCFCs)再到氫氟烴(HFCs)物質的漫長歷程。HFCs不含Cl元素,對大氣臭氧層沒有破壞,曾被認為是替代氯代烴類制冷劑的理性選擇[2]。其中,在汽車制冷行業,以1,1,1,2-四氟乙烷(CF3CFH2,HFC-134a)替代二氟二氯甲烷(CF2Cl2,CFC-12)作為典型代表。另外,1,1-二氟乙烷(CF2HCH3,HFC-152a)、三氟甲烷(CHF3,HFC-23)等氫氟烴系列的產品在混配制冷劑、發泡劑、氣霧噴射劑中有廣泛的應用,也逐漸取代高消耗臭氧潛能(ODP)的HCFCs或CFCs系列產品。所以,氫氟烴類產品曾被人們作為理想的ODS替代品而大量的生產與使用。
然而,HFCs具有很高的熱潛能效應(GWP),這是因為它能很強的吸收紅外輻射,并且大氣壽命時間很長[3-4]。HFCs和HCFCs都具有非常高的GWP,其中HFC-134a和HFC-152a的GWP分別為1 370和133,HFC-23是CO2的11 700倍,這些HFCs類物質的GWP遠遠高于CO2。《京都議定書》意識到非CO2溫室氣體影響全球氣候變暖的重要性之后,將HFCs類物質列入限排的溫室氣體行列[5]。在發展中國家,高GWP的HFC-134a主要是應用在汽車制冷領域[6-7]。
自上世紀90年代中期,隨著汽車行業的飛速發展,HFC-134a開始大量的生產與使用,目前,大氣中HFC-134a的含量仍然以每年10%的速度增長[8]。這種現狀給地球氣候環境帶來巨大的壓力,也引起國際社會的強烈關注。歐盟規定汽車制冷劑的GWP不能超過150,但是目前廣泛使用的HFC-134a汽車制冷劑已經遠遠超過歐盟規定。2010年,美國已經禁止在新上市的汽車中使用HFC-134a制冷劑,而采用新一代的2,3,3,3-四氟丙烯(HFO-1234yf)制冷劑替代,這主要是由于HFO-1234yf的ODP為0、GWP只有4,并且大氣中的壽命短[9]。2011年,歐盟開始議定禁止新汽車空調里使用HFC-134a,在2017年完成HFC-134a的淘汰[9]。
縱觀國際制冷行業的發展潮流,未來幾年,在我國汽車制冷行業,高GWP的氫氟烴類產品終將被淘汰,終究要被ODP為0、GWP小的新型氟制冷劑所取代。按照當前趨勢計算,在2050年,HFCs的排放(CO2當量)將占全球溫室氣體排放的9%~19%。
除了政府禁止HFCs產品的使用和倡導新型制冷劑的替代HFCs的手段之外,如何處理目前大量的產能過剩的HFCs系列產品或者為HFCs產品尋找下游產品的出路,這將給氟化工企業帶來巨大的考驗。據現有的報道,關于HFCs的處理方法有:氧化燃燒法、等離子法、催化水解法和催化脫氟化氫法等[10-15]。本文總結近幾十年中重要文獻報道的HFCs處理方法,將其歸納分為2大類,一是消除處理,二是資源化轉化利用。
1 HFCs的消除處理法
高GWP的HFCs類物質的減排方式主要是通過聯合國環境規劃署(UNEP)負責的清潔能源發展機制(CDM)項目,采用消除處理的方法將HFCs類物質進行結構破壞處理,轉化成CO2。消除處理法中,主要有高溫焚燒(1 200℃)、等離子法、水解法等。通過CDM項目,發達國家出資向發展中國家購買溫室氣體減排指標。根據UNFCCC提供的信息,目前全世界范圍內政府和企業已審批大約20個CDM項目用于減除HFCs;根據UNEP的統計數據,我國HF?Cs的排放量占全世界排放量的一半以上,在CDM項目中通過焚燒HFCs將減排相當于82.6 Mt的CO2。目前我國面對大量的HFCs產品,減排壓力較大。
1.1 氧化燃燒法
氧化燃燒是當今工業上成熟的技術,是一種普遍的HFCs簡單處理方法,也是目前HFCs消除的最簡單的處理方法。一般HFCs的氧化法包括以下5個過程:加熱氧化,淬火冷卻,吸收塔,中和塔,排氣處理。HFCs的氧化反應如下:

一般的氧化燃燒法是將液化石油氣(LPG)、來自風扇的燃燒空氣和HFCs廢物在燃燒器中轉化成熱煙氣(燃燒器的溫度一般高于1 200℃,停留時間是2 s)。然后煙道氣經驟冷后,大多數酸性氣體(HF和HCl)通過該冷卻過程被吸收到溶液中。驟冷后未吸收的酸性氣體和CO2、N2進入堿洗滌器,剩余的酸性氣體與CO2、N2一起進料到吸收塔。酸性氣體可以通過被堿性水吸收。將稀釋的HF溶液再循環至冷卻,其中得到濃縮的HF酸(質量分數30%~40%)。N2通過苛性堿洗滌器之后再排放到大氣中。來自吸收塔的煙霧被送到中和塔,中和塔中用苛性鈉溶液洗滌,該洗滌是為了除去酸性氣體。來自中和塔的尾氣進入排氣塔,最后排放到大氣中。
Qin等報道了一種低廉的火焰燃燒法用于HFC-134a的處理,HFC-134a、空氣摩爾比大于0.9、采用預混-散射火焰時、HFC-134a、液化氣摩爾比小于0.8時,HFC-134a降解率高達99.98%[10]。盡管該方法的降解效益高,但是依然存在副產物,如C2H2F2,CHF3和C2H3F,形成二次HFCs污染物的產生。
雖然該方法工藝簡單、技術難度不大、操作方便、效益高等優點,已經列入中國、印度、韓國、墨西哥、以色列等國家的CDM項目中,但是HFCs氧化燃燒法的一個重要缺點是分子中的氟必須作為HF的形式排出,然后從混合氣流中洗滌、處理,最終作為氟化物鹽。該方法對反應器材質的選擇還存在一定的挑戰性,該反應在1 200℃的高溫下進行,而且反應產生HF對反應器存在非常強的腐蝕性。其次,在反應從1 200℃降溫至室溫時,該過程中產生有毒物質,如二噁英,又將引起另一個環境問題。
1.2 等離子法
等離子處理是另一個已經商業化運行的工業處理路線,該技術在用于HFCs的處理方面需要非常高的溫度下進行。1992年,澳大利亞聯邦科工組織(CSIRO)和SRL Plasma公司首先聯合開發了所謂的PLASCON工藝用于氟利昂的處理,并在2007年被用于HFCs類化合物——CHF3的處理[16]。在高溫下利用PLASCON工藝可將氟利昂降解為鹵代酸(HF、HCl和HBr)和鹵素分子(F2、Cl2和Br2)。
Wofford報道表面等離子法用于CHF3,盡管消除CHF3的效益很高(達99.9%),但是產生很多有毒的物質,如COF2等[17]。有些HFCs物質,如C2F6,采用等離子法處理時會產生大量的有毒有害物質(F2、CO、COF2等)。Chang等人采用等離子和催化劑結合的方法來消除處理C2F6,很大程度上降低了F2、CO、COF2和CF4的釋放[18]。
據報道,使用水蒸汽代替O2來作為氧化劑,可以減少一些含氟的副產品[16]。水等離子法在用于HFC-134a降解時,具有非常高的降解效率[19]。水等離子通過直流電弧法產生(銅棒上嵌入鉿的陰極與噴口型的銅陽極產生直流電弧)。水等離子炬方法的優勢在于通過直流放電能產生100%的水等離子。這種獨特的生成水蒸汽法,無需其他的氣體參與,無需水猝冷步驟,該方法具有很高的能效性。HFC-134a被注入反應管內的水等離子流中進行降解反應。HFC-134a降解過程中產生的F2和HF,通過連接反應管的中和池進行吸收。這一降解過程中,通入HFC-134a氣體的流速為0~185 mmol/ min。當HFC-134a的流速與電弧功率比為0.43 mmol/kJ時,HFC-134a的降解效率達99.9%。
等離子體法盡管取得不少進步,但是仍然存在很多技術挑戰,如高溫條件、操控難等問題,而且工藝的設備投資和運行成本是一般熱焚燒法的3~5倍。HCFs中擁有寶貴的C-F鍵,利用它來合成環境友好、附加值高的其他含氟化合物是更為理想的路線。
1.3 催化水解法
HFCs的降解需要高溫,條件苛刻,一直是HFCs消除處理技術的難題。催化技術的引入大大降低HFCs的降解溫度。HFCs的降解對催化劑的高活性和催化材料的長時間耐酸性都有嚴格的要求。
Feaver等研究低含量下(質量分數3×10-3)HFC-23在ZrO2和ZrO2-SO4催化劑上的降解反應情況[20]。反應溫度為300~500℃,停留時間為0.4 s,加入H2O的質量分數為2.5%,C轉化為CO2。結果發現,在沒有水的情況下,ZrO2-SO4催化劑沒有活性,這說明HFC-23降解過程發生水解反應,而非氧化過程。在有水存在的情況下,ZrO2催化劑表面上發生的HFC-23的降解反應如下:
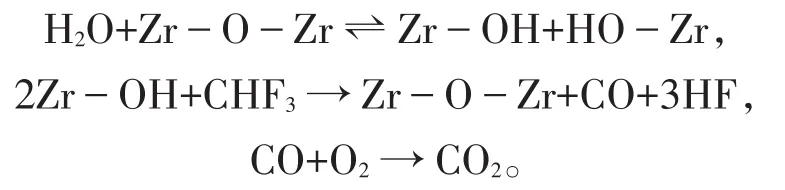
對于結構性質穩定的CF4,也是通過催化水降解的方法,采用的是AlPO4-Al2O3催化劑。Jeon等人發現水的含量對CF4降解的影響很大,而O2的幾乎沒有影響[21]。H2O對HFCs的催化降解具有很高的促進作用,原因是H2O分子吸附在催化劑的表面形成-OH基起著至關重要的作用。
在HFCs降解的反應過程中,不可避免的產生HF。HF是能與很多催化劑材料反應,破壞催化劑的活性。例如,硫酸酸化的ZrO2催化劑用于CFCs的氧化降解反應中,催化劑的失活原因就是ZrO2與HF的氟化反應導致催化劑表面的O和S失去,而且催化劑的表面積急劇下降[21]。在加入質量分數2.5%的水蒸汽后,催化劑的穩定性大大的提高。
盡管該方法催化水解技術大大降低了HFCs的降解溫度,能耗大大降低。但是,對于催化HFCs水解降解反應,催化材料不但需要較高的反應活性,而且要求很強的耐酸腐蝕環境。另外,該方法一味將HFCs中的C、F轉化為CO2和HF,其浪費豐富的CF寶貴資源,設想將HFCs轉化為含氟烯烴等高附加值產品的路線要明顯優于該方法。
2 HFCs的資源化轉化利用
上述方法對HFCs并不能進行有效的處理。直接燃燒方法具有工業化的規模,其耗能較大,是一種十分昂貴的銷毀技術,且直接燃燒過程中含氟有機廢棄物的不完全氧化將導致劇毒物質F2、COF2的生成,易造成嚴重的二次污染。等離子法和催化水解法,作為一種破壞性的處理技術,不能有效利用HFCs。
事實上,更令人感興趣的是如何開發HFCs進行資源化轉化利用,轉化成更有價值的物質,如有序碳纖維和含氟烯烴等,這樣不僅使得HFCs對全球溫室效應的作用有所降低,同時也能夠解決HFCs產品產能過剩的問題。目前,經過總結文獻資料,先將HFCs的資源化轉化利用方法主要分為高溫裂解合成含氟烯烴法、與其他烴類共裂解法和催化脫氟化氫法。
2.1 高溫裂解合成含氟烯烴
HFCs的高溫裂解法,主要是在高溫下裂解成自由的碳氟物種離子碎片,然后再自由結合形成幾種或多種反應產物。但目前高溫裂解HFCs的研究文獻較少[22]。
HFC-23高溫裂解可制得四氟乙烯(C2F4,TFE),作為HFCs高溫裂解制含氟烯烴的典型代表。HFC-23在高溫(>750℃)下可以裂解轉化為TFE,而TFE是涂料、黏結劑和聚四氟乙烯(PTFE)等工業產品的主要生產原料之一。CHF3分解總速率可以表示為5.2×1013s-1exp[(-295 kJ/mol)/(RT)),屬于中等壓力下的1級反應[23]。在低于29 kPa的壓力下,Modica等人認為CHF3裂解行為可用2級反應機制來解釋,表明CHF3裂解可能是壓力依賴性的反應。目前有關CHF3裂解制TFE的研究相對較少。在高溫下,CHF3生成TFE的反應式為:
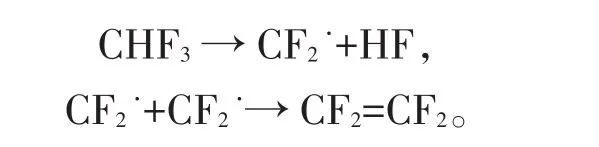
2.2 與其他烴類共裂解
HFCs與其他小分子烴類的反應,可以與甲烷、TFE、CFC-12等共裂解。該反應一般也需要較高的反應溫度,可以高效益的轉化和處理HFCs,并且能生成高附加值的工業產品或原料。
等量的HFC-32(CHF3)和CH4反應,主要的產物是偏氟乙烯(VDF)、TFE及HF,副產物包括C3F6、CH2F2、C2H3F、C2HF3、C2H6、C2H2和CHF2CHF2等。其反應式為:
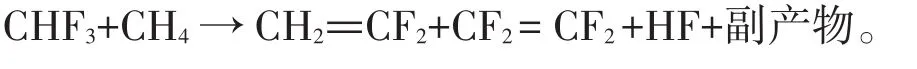
該反應的產物和CHF3的轉化率受溫度的影響很大。在低溫下,TFE和VDF的生成速率較為接近,但在高溫條件下(高于840℃),VDF逐步變為決定性的產物。在反應溫度為900℃時,CHF3的轉化率為77%,VDF的選擇性為27%,收率高達21%。同樣,該工藝路線也適用于HFCs與甲烷反應合成[24]。
Moon等報道,在反應溫度為973~1 273 K、停留時間為0.01~14 s的條件下,可以通過CHF3與C2F4的共熱解選擇性地合成六氟丙烯(C3F6,HFP)[25]。HFP主要通過CHF3熱解產生的中間體和C2F4的二聚體形成。優化反應條件如反應溫度和停留時間可顯著提高HFP的選擇性和產率。在K/活性炭催化劑存在下,單獨使用CHF3熱解,CHF3的轉化率和TFE和HFP的選擇性均明顯增強。在催化裂解過程中,推測活性炭的作用是提高關鍵中間體CF2卡賓的局部表面含量,這是吸收的CHF3消除HF后產生的。表面CF2的聚合速率提高,高于氣相反應時的聚合速率。CHF3與C2F4的共熱解反應為:
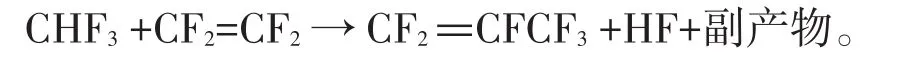
專利CN102267866A以CFC-12和HFC-134a作為原料,添加助劑的氧化鉻作為催化劑,在高溫條件下生成三氟乙烯和四氟甲烷產品[26]。該反應式為:
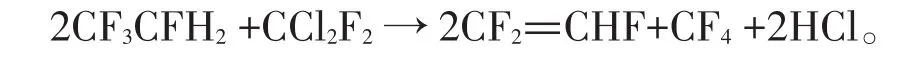
該方法的優點是,不但將高ODP和高GWP的CFC-12進行轉化為四氟甲烷產品(該產品主要用于工業微電路蝕刻),而且將高GWP的HFC-134a轉化為高附加值的三氟乙烯產品。但是,該反應的溫度高,CFC-12歧化的副反應較多,存在產物難以收集、分離和提純等問題,不適宜進行工業化。
目前,HFCs與其它小分子的烴類反應的研究,尚處于實驗室的研究階段,但是該方法反應溫度過高、反應復雜、副產物多、操控性難,而且產物分離和提純困難,偏于理想化,其實現工業化還存在很大的技術難題。
2.3 催化脫氟化氫法
HFCs脫HF反應的主要產物為含氟烯烴,制備含氟烯烴具有重要的經濟和環境價值。含氟烯烴可用作含氟聚合物的單體和含氟精細化學品的中間體,如合成聚氟乙烯的氟乙烯、合成三氟溴乙烯的三氟乙烯等[27]。另外,某些含氟烯烴ODP為0、GWP極低,對全球溫室效應影響小,被認為是新一代環保型制冷劑,如1,3,3,3-四氟丙烯(HFO-1234ze)、HFO-1234yf等。
在制備含氟烯烴的制備路線中,其中HFCs脫氟化氫合成含氟烯烴是一簡便、高效的方法,是氟化工界研究的熱點之一。然而,HFCs脫氟化氫在熱力學和動力學上受限制,所以有必要采用高效的催化劑,降低反應溫度,提高反應的速度[28-29]。目前關于HFCs脫HF的反應研究,主要集中在C2和C3的氫氟烴中,主要有HFC-152a脫HF生成氟乙烯(VF),1,1, 1-三氟乙烷(HFC-143a)脫HF生成VDF,HFC-134a脫HF生產三氟乙烯(TrFE),1,1,1,3,3-五氟丙烷(HFC-245fa)脫HF制備HFO-1234ze,1,1,1,2,3,3-六氟丙烷(HFC-236ea)脫HF制備1,1,1,2,3-五氟丙烯(HFO-1225ye)等。
HFC-152a脫HF是生成VF的一種重要方法[30]。VF作為聚氟乙烯(PVF)的基本原料,還可以用作其他共聚物的單體。但反應中存在副產品HF,后期循環利用時,耗費大量人力和財力。為了避免上述缺陷和提高VF的收率,采用通入乙炔與HFC-152a一起進行脫HF反應。Al2O3-Fe2O3催化劑和添加助劑的Al2O3-Fe2O33組份催化劑對該反應體系,都有較高的活性和選擇性,其中后者的性能更佳。
HFC-143a作為生產HFC-134a過程中產生一種的副產品,其利用價值不大,目前尚未發現其工業用途[31-32]。所以,將大量的副產品HFC-143a進行脫HF生產VDF是很有必要的,而VDF是一種含氟聚合物的重要原料。Li等人采用催化脫HF的方法用于HFC-143a轉化VDF,結果發現,具有弱酸性的Mg2P2O7催化劑具有較為穩定的活性和高的VDF選擇性,在100 h反應后,轉化率維持在50%左右,選擇性達95%[31-32]。
HFC-134a脫HF的方法研究文獻報道較少,主要是專利。蔚等報道了HFC-134a脫HF制備三氟乙烯的反應情況,以添加金屬助劑Fe、Mg、Y和Zn的氧化鋁作為催化劑,結果發現,以沉積沉淀法制備的Mg/AlF3催化劑具有較高的催化劑性能,450℃時三氟乙烯的收率可達35%,三氟乙烯選擇性大于99%[33]。FR2710054A報道了一種三氟乙烯的制備方法[13]。使用氟化鋁作為HFC-134a脫HF反應的催化劑;US5856593A是采用摻雜其它金屬的鉻氧化物作為HFC-134a脫HF制備三氟乙烯的高效催化劑[15];FR2729136A報道HFC-134a脫HF制三氟乙烯時,通入BF3來提高催化劑的活性[14]。
由此可見,關鍵技術的核心還是在于高效催化劑的開發。該工藝經催化脫HF反應來制備三氟乙烯,HFC-134a是大宗化學品,原料成本低,該方法具有反應路線簡單、催化劑成本低和三氟乙烯選擇性高等特點,易于產物收集。
Mukhopadhyay等研究了1,1,1,2,2-五氟丙烷(HFC-245cb)氣相催化脫HF制備HFO-1234yf,采用活性炭或活性炭負載貴金屬作為脫HF反應的催化劑,其中發現0.5%的Pd/活性炭催化劑顯示出較好的性能[34]。Miller等采用活性炭為HFC-245cb脫HF的催化劑,產物主要以HFO-1234yf為主,轉化率為23.6%,選擇性高達97%;當采用活性炭作為HFC-245fa脫HF的催化劑時,轉化率為38%,HFO-1234ze的選擇性達96%[35]。Hirokazu等研究了催化HFC-236ea制備HFO-1225ye的反應性能,采用氟化的氧化鉻和氧化鉻為催化劑,結果發現,氟化后的氧化鉻性能優于未經氟化的氧化鉻,氟化處理可提高催化劑的反應性能[36]。
更多的科研工作者對催化劑進行處理,在轉化率和產品選擇性方面都有較大的提升,然而對于C3系列的HFCs脫HF反應工藝中,催化劑都存在短時間內容易失活的共同弱點。
HFCs催化脫氟化氫生成高附加值的含氟烯烴的方法,屬于原子經濟反應,具有操作易控、分離提純容易、腐蝕小等優點,而且大部分含氟烯烴是目前氟化工重要含氟聚合物的原料。該方法尤其對HFC-134a的資源轉化是一個理想的選擇。對于目前許多生產HFC-134a的企業,將直接找到下游三氟乙烯產品的出路,這種方法能直接利用現有的成熟HFC-134a化工工藝設備,對于我國的氟化工二次產業結構升級具有非常重要的意義。
3 結語與展望
HFCs產品曾認為是消耗臭氧層物質CFCs和HCFCs的理想替代品,被廣泛應用于冰箱、空調等產品的制冷劑中,盡管其ODP為0,對臭氧層沒有破壞作用,卻具有極高GWP。隨著CFCs和HCFCs物質在《蒙特利爾議定書》框架下加速淘汰,全球HFCs產品的生產與消費量正在快速上升,HFCs被認為是目前世界上增長最快的溫室氣體,其排放量正以每年10%的速率增加。根據聯合國環境規劃署臭氧秘書處科學評估小組的報告,到2050年HFCs對于全球CO2排放的貢獻率將達到25%,這將對全球氣候變化造成不可估量的影響,嚴重威脅人們的生存環境。
氫氟烴類化合物HFCs作為非CO2類溫室效應氣體,含有寶貴的C-F鍵,利用高溫氧化燃燒或等離子來破壞C-F鍵,從能量和資源利用角度來說均不是理想的選擇。目前,《蒙特利爾議定書》締約國就HFCs減排的問題達成一致協議。盡管利用HFCs焚燒進行溫室氣體減排可以得到碳產品,符合聯合國CDM項目的碳交易計劃,我國可以獲得一定的碳交易補貼。但是,由于經濟原因,碳交易價格逐年下跌,加上其他不穩定因素,依靠國外碳交易補貼進行HFCs焚燒減排溫室氣體難以長期維系。因此,盡早開展HFCs減排,特別是其資源化利用是十分迫切和必要的。
HFCs類物質可以轉化為附加值高、環境友好的含氟烯烴、含氟聚合物和碘氟烴等,如TFE、HFP、VDF、CF3I等。這也是未來HFCs的資源化轉化利用的意義所在。雖然利用HFCs裂解或與其他烴類共裂解合成含氟烯烴單體,需要的溫度相對較高,收率偏低,難以與現有的含氟烯烴工業生產路線競爭,但是在高效催化劑作用下,反應溫度可以大幅度降低,并且含氟烯烴的收率可以明顯提高。因此,催化脫HF反應路線的技術關鍵問題是高效催化劑的開發。
[1]Molina M J,Rowland F S.Stratospheric sink for chlorofluoromethanes:chlorine atomc-atalysed destruction of ozone[J]. Nature,1974,249:810-812.
[2]何軍.氣相氟化合成HFC-134a的CrOx-Y2O3催化劑的表征與性能研究[D].金華:浙江師范大學,2008.
[3]Sihra K,Hurley M D,Shine K P,et al.Updated radiative forcing estimates of 65 halocarbons and nonmethane hydrocarbons[J].Journal of Geophysical Research:Atmospheres, 2001,106(D17):20493-20505.
[4]Piers M D F,Joshi M.The role of halocarbons in the climate change of the troposphere and stratosphere[J].Climatic Change,2005,71(1/2):249-266.
[5]Shine K P,Sturges W T.Atmospheric science.CO2is not the only gas[J].Science,2007,315(5820):1804-1805.
[6]McCulloch A,Midgley P M,Ashford P.Releases of refrigerant gases(CFC-12,HCFC-22 and HFC-134a)to the atmosphere[J].Atmospheric Environment,2003,37(7):889-902.
[7]Papasavva S,Luecken D J,Waterland R L,et al.Estimated 2017 refrigerant emissions of 2,3,3,3-tetrafluoropropene (HFC-1234yf)in the United States resulting from automobile air conditioning.Environmental science&technology,2009, 43(24):9252-9259.
[8]Hofman D J,Butler J H,Dlugokencky E J,et al.The role of carbon dioxide in climate forcing from 1979 to 2004:intro-duction of the Annual Greenhouse Gas Index[J].Tellus B, 2006,58(5):614-619.
[9]Henne S,Shallcross D E,Reimann S,et al.Future emissions and atmospheric fate of HFC-1234yf from mobile air conditioners in Europe.Environmental science&technology,2012, 46(3):1650-1658.
[10]Qin L,Han J,Liu L,et al.Highly efficient decomposition of HFC-134a by combustion oxidization method[J].Fresenius Environmental Bulletin,2013,22(7):1919-1923.
[11]黃超群,楊斌,楊銳,等.氟里昂134a的真空紫外光電離和光解離的同步輻射研究[J].高等學校化學學報,2005,26 (12):2314-2318.
[12]Saito H,Watanabe T.Decomposition mechanism of fluorinated compounds in water plasmas generated under atmospheric pressure[J].Plasma Chemistry and Plasma Processing, 2010,30(6):813-829.
[13]Serge H,Andre L.Catalystsforpreparation oftrifluoroethylene by dehydrofluorination of tetrafluoroethane:FR2710054[P]. 1995-03-24.
[14]Serge H.Process for dehydrofluorinating fluoroalkanes to fluoroalkenes:FR2729136[P].1997-02-07.
[15]Powell R L,Sharratt A P.Process for the production of fluorine containing olefins:US5856593[P].1999-01-05.
[16]Murphy A B,Farmer A J D,Horrigan E C,et al.Plasma destruction of ozone depleting substances[J].Plasma Chemistry and Plasma Processing,2002,22(3):371-385.
[17]Wofford B A,Jackson M W,Hartz C,et al.Surface wave plasma abatement of CHF3and CF4containing semiconductor process emissions[J].Environmental science&technology,1999,33(11):1892-1897.
[18]Chang M B,Lee H M.Abatement of perfluorocarbons with combined plasma catalysis in atmospheric-pressure environment[J].Catalysis Today,2004,89(1/2):109-115.
[19]Watanabe T,Tsuru T.Water plasma generation under atmospheric pressure for HFC destruction[J].Thin Solid Films, 2008,516(13):4391-4396.
[20]Feaver W B,Rossin J A.The catalytic decomposition of CHF3over ZrO2-SO4[J].Catalysis today,1999,54(1):13-22.
[21]Jeon J Y,Xu X F,Choi M H,et al.Hydrolytic decomposition of PFCs over AlPO4-Al2O3catalyst[J].Chemical Communications,2003(11):1244-1245.
[22]Tschuikow-Roux E,Marte J E.Thermal Decomposition of Fluoroform in a Single-Pulse Shock Tube.I.Journal of Chemical Physics[J].1965,42:2049-2056.
[23]Han W F,Kennedy E M,Kundu S,et al.Experimental and Chemical Kinetic Study of the Pyrolysis of Trifluoromethane and the Reaction ofTrifluoromethanewith Methane[J].Journal of Fluorine Chemistry,2010,131(7):752-761.
[24]Yu H,Kennedy E M,Mackie J C,et al.An experimental and kinetic modeling study of the reaction of CHF3with methane[J].Environmental Science&Technology,2006,40 (18):5778-5785.
[25]Moon D J,Chung M J,Kim H,et al.Pyrolysis of Trifluoromethane to Produce Hexafluoropropylene[J].Industrial and Engineering Chemistry Research,2002,41:2895-2902.
[26]羅孟飛,蔚辰剛,賈文志,等.一種三氟乙烯和四氟甲烷的制備方法,2011101155627[P].2011-05-05.
[27]白彥波,馬洋博,毛偉,等.氣相催化脫氟化氫制備含氟烯烴催化劑的研究進展[J].化工進展,2013,32(10):2387-2391.
[28]Baker A W,Bonniface D,Klap?tke T M,et al.Catalytic fluorination of trichloroethene by anhydrous hydrogen fluoride in the presence of fluorinated chromia under static conditions.Synthesis of[18F]-labelled CF3CH2F and[36Cl] -labelled CF3CH2Cl.Catalytic dehydrofluorination of CF3CH 2F and CF3CH2Cl[J].Journal of Fluorine Chemistry,2000,102 (1):279-284.
[29]Kohne A,Kemnitz E.Heterogeneous catalyzed synthesis of 1,1,1,2-tetrafluoroethane from 1,1,1,2-tetrachloroethanethermodynamicsand reaction pathways[J].JournalofFluorine Chemistry,1995,75(1):103-110.
[30]Sirlibaev T S,Akramkhodzhaev A,Usmanov K U.Catalytic synthesis of vinyl fluoride from 1,1-difluoroethane and acetylene[J].Journal of Applied Chemistry of the USSR, 1985,58(7):1541-1543.
[31]Li G L,Nishiguchi H,Ishihara T,et al.Catalytic dehydrofluorination ofCF3CH3(HFC-143a)into CF2CH2(HFC-1132a) [J].Applied Catalysis B:Environmental,1998,16(4):309-317.
[32]Li G L,Ishihara T,Nishiguchi H,et al.Novel Catalysts Effective for Dehydrofluorination of CF3CH3(HFC143a)into CF2CH2[J].Chemistry Letters,1996(7):507-508.
[33]蔚辰剛,謝冠群,周強,等.MgF2-AlF3催化劑用于四氟乙烷裂解制備三氟乙烯[J].工業催化,2012,20(4):56-59.
[34]Mukhopadhyay S,Tung H S,Puy M V D,et al.Method for producing fluorinated organic compounds:US20070197841 [P].2007-08-23.
[35]Miller R N,Nappa M J,Rao V N M,et al.Azeotrope compositions comprising 2,3,3,3-tetrafluoropropene and hydrogen fluoride and uses thereof:US7476771[P].2009-01-13.
[36]Hirokazu A,Eiji S.Method for manufacturing 1,1,1,2,3-pentafluoropropene and method for manufacturing 1,1,1,2, 3-pentafluoropropane:EP0726243[P].1996-08-14.
TQ246.31
ADOI10.3969/j.issn.1006-6829.2016.04.001
上海市科學技術委員會項目(15dz1181000),湖北理工學院人才引進項目(16xjz05R),大學生科技創新項目(16cx03,17cx11),國家級大學生創新創業訓練計劃項目(201610920008),湖北理工學院實驗室開發基金(201617513)
*通訊聯系人。電子郵件:zhuzhirong@tongji.edu.cn
2016-05-06
猜你喜歡
兒童故事畫報(2019年5期)2019-05-26 14:26:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
Coco薇(2016年2期)2016-03-22 02:42:52
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
