鉑催化吲哚丙炔酯合成咔唑類化合物*
2016-06-05 08:13:51張寧王德偉
化工科技 2016年3期
關鍵詞:催化劑
石 赟,張寧,王德偉
(蘭州大學 化學化工學院,甘肅 蘭州 730000)
芳環和雜環化合物在有機合成和藥物合成中占有主導性地位。因而如何高效地設計和合成此類化合物始終是有機化學工作者所面臨的挑戰之一。咔唑類化合物作為重要的含氮雜環化合物,是煤焦油中經濟價值最高的成分之一。此結構在具有生物活性的雜環化合物合成中具有重要的作用。2009年,Benjamin J Stokes小組以二聯芳基疊氮為原料,在Rh2(Ⅱ)的催化作用下合成咔唑類化合物[1]。2014年,Kazutaka Takamatsu小組又以二氨基聯苯為原料,通過Cu(OAc)2催化分子內C—H/N—H偶聯來合成咔唑[2]。2015年,Michael J James小組以3-炔醇吲哚為原料,以Ag(Ⅰ)為催化劑成功合成了咔唑類化合物[3]。
作者以N-甲基-3-吲哚丙炔酯為反應底物,討論這類反應的可能性。考察了不同過渡金屬催化劑對反應的影響,并探討了反應溶劑、溫度、反應時間對該反應分離產率的影響。
1 實驗部分
1.1 試劑與儀器
氫氧化鉀、二甲基亞砜(DMSO)、N,N-二甲基甲酰胺、無水乙醚、四氫呋喃、甲苯、苯甲醚、1,4-二氧六環、乙腈、1,2-二氯乙烷、二氯甲烷:分析純,利安隆博華(天津)醫藥化學有限公司;吡啶:分析純,上海中泰化學試劑有限公司。
實驗中未經說明的藥品和溶劑均為市售分析純試劑,所有反應均用薄層硅膠板(GF254)進行TLC監測跟蹤,產物利用硅膠柱純化(200~300 μm),部分硅膠柱需要用三乙胺壓洗,所用洗脫劑為工業乙酸乙酯與工業石油醚(沸程60~90 ℃)的混合液。
1H NMR和13C NMR使用AM-400核磁共振波譜儀:溶劑選用CDCl3,內標為TMS,德國Bruker公司;質譜使用[70eV(EI)]質譜儀:ZAB-HS,英國VG公司。
1.2 吲哚丙炔酯的制備
1.2.1 氮保護基吲哚的制備
在50 mL的圓底燒瓶中加入相應的吲哚(5.0 mmol,0.586 g)和KOH(10.0 mmol,0.560 g),然后加入20 mL的DMSO,攪拌,再加入碘甲烷(10.0 mmol,1.420 g),室溫下反應。TLC監測反應結束后,用20 mL水淬滅反應,用乙酸乙酯萃取3次,合并有機相,無水硫酸鈉干燥,減壓除去溶劑,粗產品柱色譜分離,得0.69 g黃褐色固體,產率100%[4],反應方程式如下。

1.2.2 N-甲基-3-吲哚甲醛的制備
在100 mL的具支燒瓶中加入無水甲苯(10 mL),在氬氣條件下滴加N,N-二甲基甲酰胺(4 mmol,0.330 g)。冷卻至0~-5 ℃,緩慢滴加三氯氧磷(0.5 mL),滴畢,在室溫下攪拌30 min,將N-甲基吲哚(5 mmol)溶于4 mL無水甲苯中,在惰氣條件下緩慢滴加。然后在室溫下反應3 h,加入4 mol/L NaOH溶液60 mL,加熱回流,TLC檢測,冷至室溫,乙酸乙酯萃取3次,合并有機相,無水硫酸鈉干燥,減壓蒸餾,粗產品柱色譜分離,得0.443 2 g的黃褐色固體,產率59%[5],反應方程式如下。
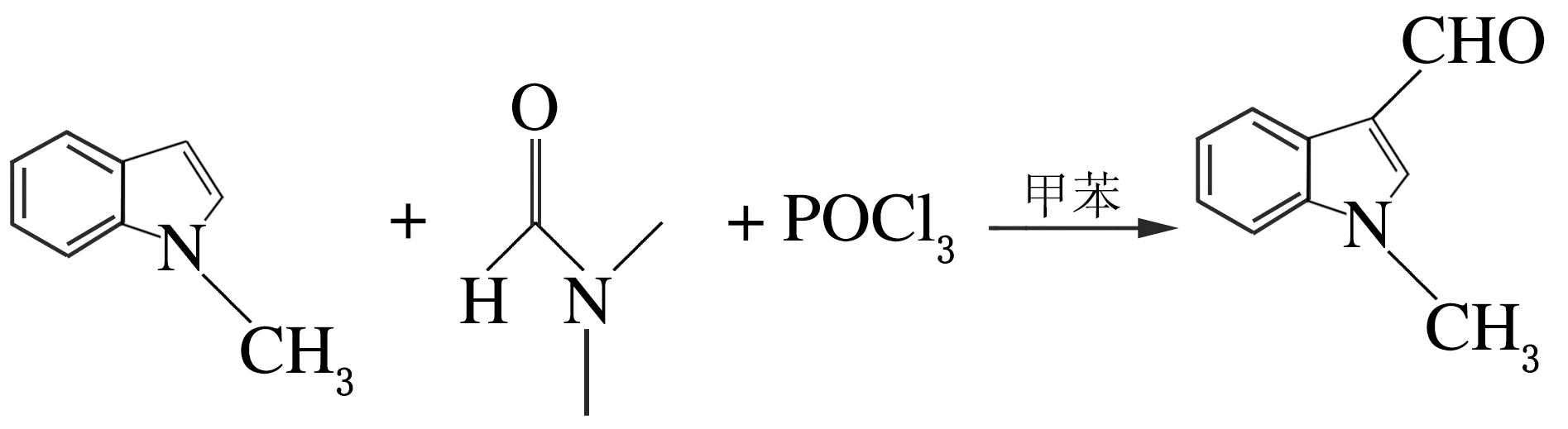
1.2.3 N-甲基-3-吲哚丙炔醇的制備
氬氣保護下,在50 mL的具支燒瓶中加入鎂粉(5 mmol,0.120 g)和二氯化汞(0.05 mmol,0.014 g),加入無水乙醚做溶劑(5 mL),攪拌,緩慢滴加炔丙基溴(5 mmol,0.595 g)溶于無水乙醚(3 mL)的溶液,室溫下反應1 h,直至反應液由黃色變為灰色。在冰鹽浴條件下,把N-甲基-3-吲哚甲醛(2.5 mmol,0.403 g)溶于無水四氫呋喃(3 mL),在氬氣下緩慢加入燒瓶中,反應1 h。反應結束后,用飽和氯化銨溶液將反應淬滅,乙酸乙酯萃取3次,合并有機相,硫酸鎂干燥,減壓蒸餾除去溶劑,快速柱色譜分離,即可得3-吲哚炔醇,黃色液體,產率62%[6],反應方程式如下。

1.2.4 N-甲基-3-吲哚丙炔酯的制備
在冰浴下,N-甲基-3-吲哚丙炔醇(1.5 mmol,0.302 g),吡啶(0.4 mL)和4-二甲氨基吡啶(0.33 mmol,40mg)加入50 mL圓底燒瓶中,緩慢加入醋酸酐(0.32 mL),反應6 h,加入飽和碳酸氫鈉溶液(20 mL),二氯甲烷萃取3次,合并有機相,硫酸鎂干燥,減壓蒸餾除去溶劑,柱色譜分離即可得3-吲哚丙炔酯,產率91%[6],反應方程式如下。

1.3 N-甲基咔唑的制備合成
在10 mL的反應管中,依次加入稱量好的N-甲基-3-吲哚丙炔酯(0.072 3 g,0.3 mmol),催化劑PtCl2[n(PtCl2)∶n(N-甲基-3-吲哚丙炔酯)=10%],然后加入甲苯溶液(4 mL),置于120 ℃的油浴中,維持此溫度,攪拌反應4 h,TLC監測反應的進程。當反應完全后,停止加熱,冷卻至室溫,用水淬滅,倒入分液漏斗中,用乙酸乙酯溶液萃取3次,然后合并有機相,用飽和的NaCl溶液洗滌反萃有機相,用硫酸鎂干燥,減壓蒸餾除去溶劑,柱色譜分離即可得到N-甲基咔唑。
2 結果與討論
在實驗的初始階段,構建合成了化合物N-甲基-3-吲哚丙炔酯(1a),以其為底物討論Pt催化吲哚丙炔酯反應的可能性,并對反應條件進行了篩選,反應方程式如下。
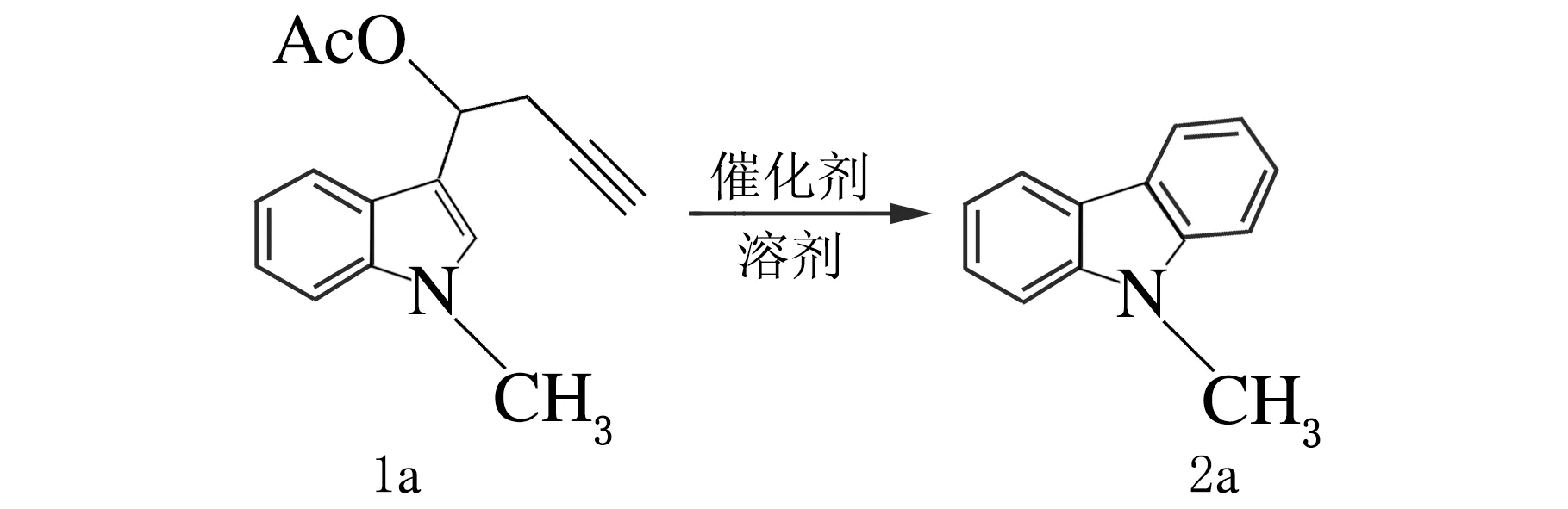
2.1 催化劑種類和溶劑對分離產率的影響
實驗以1a為例進行了催化劑和溶劑的選擇。考慮到空白實驗的重要性,先進行了空白實驗操作,在不添加任何催化劑的條件進行了實驗,然后又選用了Pt(MeCN)2Cl2、 PtCl2、PtBr2、PtCl4和PtI4等不同的Pt催化劑[n(Pt催化劑)∶n(1 a) =10%]來進行嘗試。對于大多數有機反應來說,溶劑的選擇也非常重要,所以實驗分別選用了甲苯、苯甲醚、1,2-二氯乙烷(DCE)、乙腈(MeCN)、1,4-二氧六環、二氯甲烷(DCM)等不同溶劑進行反應,以尋找具有最佳效果的溶劑,結果見表1。
研究發現在不添加催化劑的條件下沒有產物生成,但加入Pt的催化劑均有產物生成,說明Pt催化劑都能催化這類環化反應的發生,其中PtCl2的催化效率最高,產率可達到67%。選擇溶劑的實驗表明,各種不同溶劑甲苯、苯甲醚、DCE、MeCN、1,4-二氧六環、DCM等對反應都有影響。而且,在PtCl2的催化下,這幾種溶劑也都能使底物發生成環反應,用甲苯作溶劑時反應進行得最好。

表1 催化劑種類以及溶劑對分離產率的影響1)
1) 反應條件:1a(0.3 mmol),n(Pt催化劑)∶n(1a) =10%,溶劑4 mL,溫度120 ℃,反應時間4 h。
2.2 溫度與時間對分離產率的影響
隨著所用催化劑、溶劑的確定,需要對反應溫度與時間進行優化,所以進行了以PtCl2為催化劑、甲苯為溶劑,采用不同的溫度與時間的實驗,以尋找具有最佳效果的反應溫度與時間,實驗結果見表2。

表2 反應溫度與時間對分離產率的影響
從表2可以看出,反應的最佳溫度為120 ℃,低于120 ℃,產物的收率明顯降低。而最適宜的反應時間為4 h,反應時間減短,產率有所降低,反應時間增長,產率并沒有增加。
2.3 底物的拓展
經過對催化劑、溶劑、反應溫度和時間的篩選,確定了n(PtCl2)∶n(1a) =10%,甲苯作溶劑,120 ℃條件下反應4 h,所生成的環化產物的產率最高可達67%,反應方程式如下。在得出最優反應條件之后,對該環化反應的底物適用范圍進行了探討,結果見表3。
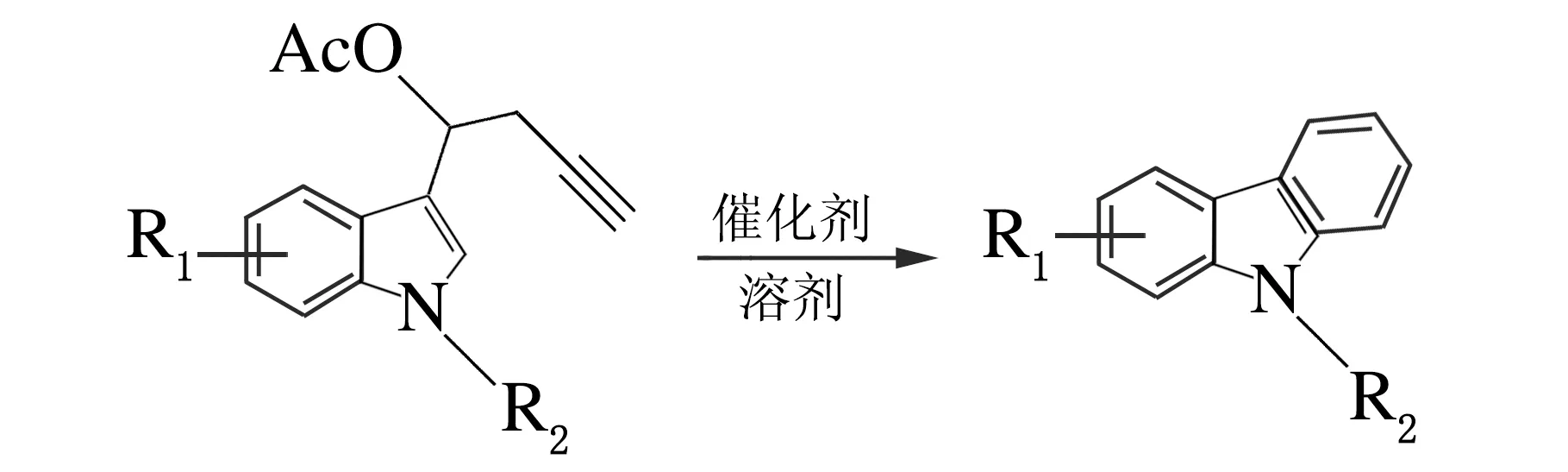
表3 底物的拓展1)

序號底物Ⅰ產物 Ⅱ產率/%1671a2a2<51b[5-7]2b3801c[5-6,8]2c4621d[4-6]2d5721e[5-6,8]2e6641f[4-6]2f7461g[4-6]2g
1) 反應條件:底物Ⅰ0.3 mmol,n(Pt催化劑)∶n(底物Ⅰ) =10%,溶劑4 mL,反應溫度120 ℃,反應時間4 h。
表3合成了7個吲哚丙炔酯類化合物來拓展底物,發現吲哚上不同的取代基,吲哚N上不同的保護基團,都會對反應的產率產生一定的影響。吲哚上取代基的影響相對較小,而吲哚N上不同保護基團對反應影響較大,且發現保護基團的吸電子性越強,成環反應越容易進行、產率越高。
2.4 反應機理的推測
在查閱了一定的文獻資料[9-10],并對其進行分析研究后,結合實驗研究結果,通過相關的對照實驗,提出了PtCl2參與的可能反應機理。首先是乙酰基的羰基進攻經金屬鉑活化過的炔基,形成反應中間體Ⅲ,接著羰基氧上的電子向炔基轉移,形成環狀過渡態金屬卡賓中間體Ⅳ,隨后炔基烯丙位的C—O斷裂,電子重新排布,進一步與吲哚2位環化形成六元環,最后脫除一分子乙酸得到目標產物,反應方程式如下。
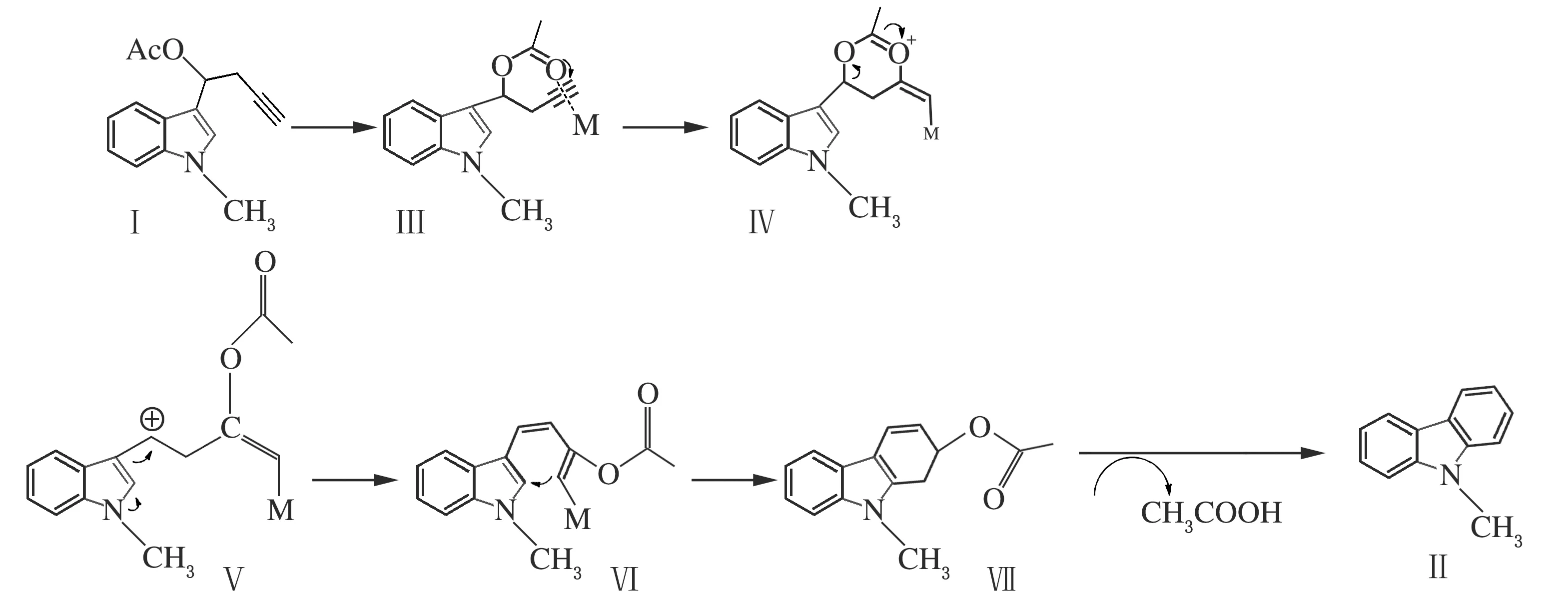
圖1 可能的反應機理
3 結 論
實現了用吲哚丙炔酯在PtCl2催化條件下分子內成環生成咔唑的反應,以氯化鉑為催化劑,甲苯做溶劑,120 ℃下反應4 h,可以達到67%的產率。反應過程簡單,后處理方便,且底物適用性較好,是一種新的合成咔唑衍生物的實驗方法,為其在藥物、材料等領域的應用提供了新的合成思路。
參 考 文 獻:
[1] BENJAMIN J STOKES,BRANKICA JOVANOVIC,DONG HUIJUN,et al.Rh2(Ⅱ)-catalyzed synthesis of carbazoles from biaryl azides[J].J Org Chem,2009,74 (8):3225-3228.
[2] KAZUTAKA TAKAMATSU,KOJI HIRANO,et al.Synthesis of carbazoles by copper-catalyzed intramolecular C—H/N—H coupling[J].Org Lett,2014,16 (11):2892-2895.
[3] MICHAEL J JAMES,ROSA E CLUBLEY,KLEOPAS Y PALATE,et al.Silver(Ⅰ)-catalyzed dearomatization of alkyne-tethered indoles:divergent synthesis of spirocyclic indolenines and carbazoles[J].Org Lett,2015,17(17):4372-4375.
[4] ZHANG L,PENG C,ZHAO D,et al.Cu(Ⅱ)-catalyzed C—H (SP3) oxidation and C—N cleavage:base-switched methylenation and formylation using tetramethylethylenediamine as a carbon source[J].Chem Commun,2012,48:5928-5930.
[5] BAGHER AMIR-HEIDARI,JENNY THIRLWAY,JASON MICKLEFIELD.Stereochemical course of tryptophan dehydrogenation during biosynthesis of the calcium-dependent lipopeptide antibiotics[J].Org Lett,2007,9(8):1513-1516.
[6] XU M,REN T T,LI C Y.Gold-catalyzed oxidative rearrangement of homopropargylic ether via oxonium ylide[J].Org Lett,2012,14(18):4902-4905.
[7] KEIGO KAMATA,JUN KASAI,KAZUYA YAMAGUCH,et al.Efficient heterogeneous oxidation of alkylarenes with molecular oxygen[J].Org Lett,2004,6(20):3577-3580.
[8] YU YOSHII,TAKANORI OTSU,NORIHIKO HOSOKAWA,et al.Synthetic studies toward penitrem E:enantiocontrolled construction of B-E rings[J].Chem Commun,2015,51:1070-1073.
[9] SANDRO CACCHI,GIANCARLO FABRIZI,et al.Palladium-catalyzed synthesis of 2-(aminomethyl) indoles from 3-(o-trifluoroacetamidoaryl)-1-propargylic alcohols and amines[J].J Org Chem,2014,79(1):401-407.
[10] ILARIA AMBROGIO,SANDRO CACCHI,GIANCARLO FABRIZI.Palladium-catalyzed synthesis of 2-(aminomethyl)indoles from ethyl 3-(o-trifluoroacetamidophenyl)-1-propargyl carbonate[J].Org Lett,2006,8(10):2083-2086.
Abstract: Carbazole compounds were synthesized by indole propargylic esters in the presence of PtCl2in toluene at 120 ℃ for 4 h.The chemical structures of these obtained compounds were characterized by MS,1H NMR and13C NMR.Based on the study of the reaction mechanism and influencing factors,we found that the N of indole bearing stronger electron-withdrawing groups participated well in the cyclization reaction.
Keywords: Indole propargylic esters;Carbazole;Transition metal catalysis;Cyclization reaction
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
